










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.27 n.1 Caracas jun. 2008
Insulinorresistencia e hiperinsulinemia como factores de riesgo para enfermedad cardiovascular
Joselyn Rojas; Valmore Bermúdez; Elliuz Leal; Raquel Cano, Yettana Luti, Luis Acosta; Freddy Finol; Daniel Aparicio, Nailet Arraiz, Sergia Linares, Edward Rojas, Roger Canelón, Sánchez Deisiree.
Centro de Investigaciones Endocrino-Metabólicas Dr. Félix Gómez. Facultad de Medicina. La Universidad del Zulia. Maracaibo-Venezuela e-Mail: vbermudez@hotmail.com
Resumen
La Insulinoresistencia se define como un estado metabólico en el cual los efectos periféricos titulares de la insulina se encuentran disminuidos. La resistencia a la acción de esta hormona se compensa mediante un aumento en su secreción por parte de la célula β, resultando en la llamada hiperinsulinemia compensadora. Desde hace varios años se ha acumulado suficiente evidencia de que la insulinoresistencia y la hiperinsulinemia están involucradas en el desarrollo de hipertensión arterial, obesidad y diabetes. Igualmente, la hiperinsulinemia esta altamente relacionada con el desarrollo de de dislipidemia caracterizada por aumento de las VLDL y TAG y una disminución de las HDL favoreciendo la aparición de ateroesclerosis. Otra de las patologías que se ha encontrado fuertemente relacionada con la hiperinsulinemia y la insulinoresistencia es la isquemia miocárdica, tanto en su génesis como en su evolución, ya que se ha demostrado que las posibilidades de supervivencia del miocito se ven reducidas por la disminución de la captación de glucosa durante el período isquémico.
La hiperinsulinemia también se relaciona con la hipertrofia miocárdica, probablemente debido al efecto directo de la insulina sobre la elevación de la presión arterial, bien por incremento en la reabsorción de Na+ o por hiperactividad simpática.
Finalmente, la resistencia a la insulina es muy prevalente en pacientes no diabéticos que han padecido TIA o ACV sin secuelas. Este hallazgo tiene importantes implicaciones terapéuticas si el tratamiento de esta condición es capaz de reducir la prevalencia de enfermedad cerebro-vascular y enfermedad coronaria.
Palabras clave: insulinoresistencia, hiperinsulinemia, insulina, enfermedad cardiovascular
Abstract
Insulin resistance is defined as a metabolic state in which insulin peripheral effects are diminished. This condition is compensated by an insulin secretion increase called compensatory hyperinsulinemia. Several years ago several authors are agree with the fact that insulin resistance and hiperinsulinemia are involved in hypertension, obesity and diabetes. Equally, hyperinsulinemia is related with a plasmatic lipidic pattern characterized by a decrease in HDL cholesterol and increases in triglyceride and VLDL levels that in turn conduces to atherosclerosis development. In this sense, myocardial ischemia has been related with these conditions in both, genesis and further evolution because accelerated atherosclerosis and myocyte survival reduction by angiogenesis blockade at insulin-signalling level.
Hyperinsulinemia is related with myocardial hypertrophy. One hypothesis that has been designed to explain this association is that insulin may directly increase blood pressure and therefore left ventricular work. In support of this, insulin has been shown to activate the sympathetic nervous system in patients with essential hypertension.
Finally, impaired insulin sensitivity is highly prevalent among non-diabetic patients with a recent TIA or non-disabling ischemic stroke. This finding has important therapeutic implications if treatment to improve insulin sensitivity is shown to reduce risk for subsequent stroke and heart disease.
Key words: insulin resistance, hiperinsulinemia, insulin, cardiovascular disease.
Recibido: 06/01/2008 Aceptado: 05/03/2008
Introducción
La insulinorresistencia1 (IR) se define como el estado metabólico en el cual los efectos tisulares de la insulina se encuentran disminuidos. El término se aplica desde 1936, cuando Himsworth y colaboradores2 describieron los diferentes rangos de sensibilidad para la acción de esta hormona. La insulinorresistencia incluye defectos en las acciones metabólicas y no metabólicas de la insulina, tales como la homeostasis de la glucosa, de los lípidos y de las proteínas, efectos mitógenos, diferenciación celular, las modificaciones electrofisiológicas cardíacas y la regulación del tono arterial.
La resistencia a la acción de ésta hormona anabólica, se compensa con un aumento en su secreción por parte de la célula β pancreática, de lo cual resulta hiperinsulinemia, que prolonga el estado de IR fundamentalmente por regulación en bajada de sus receptores.
Señalización insulínica
La Insulina y su receptor
La insulina es una hormona proteica de 51 aminoácidos (aa), con un peso de 5.800 kDa, constituida por dos cadenas, una α de 21aa y una β de 30aa. Ambas cadenas están unidas mediante dos puentes disulfuro (Cis7 – Cis6; Cis19 – Cis20), y un puente disulfuro intracatenario entre las cadena α (Cis11– Cis6). El receptor de Insulina3 (RI) es un complejo heterotetramérico, constituido por cuatro cadenas, 2 α y 2 β, con un peso molecular total de 480 kDa. Las cadenas α son totalmente extracelulares y sirven como anclaje a la insulina mediante regiones ricas en cisteína (Cis524, Cis682, Cis683, Cis685), que además son reguladoras de la función catalítica de las cadenas β, las cuales son extra, trans e intracelulares, y constan de cuatro dominios: 1) Un dominio transmembrana que sirve de anclaje, el cual contiene aa hidrofóbicos en forma de α hélice. 2) Un dominio juxtamembrana, que sirve para la internalización del receptor. 3) Un dominio con capacidad catalítica tipo tirosincinasa, que presenta tres residuos tirosina (Tir1158, Tir1162, Tir1163), autofosforilables, más un residuo de lisina (Lis1018), capaz de unir al ATP. 4) Un dominio carboxilo terminal, el cual contiene los residuos de serina y treonina (Ser1294, Ser1315 y Tre1336), que sirven de reguladores junto a dos residuos de tirosina (Tir1376 y Tir1388), igualmente capaces de autofosforilarse.
Al unirse la insulina al receptor se activa su propiedad intrínseca de tirosincinasa de autofosforilación de los residuos de tirosina de las cadenas β, lo cual prepara al receptor para iniciar la cascada de fosforilación4,5. Para ello utiliza los residuos de tirosina ya fosforilados como sitios de anclaje. Las primeras moléculas en interactuar con el receptor es el IRS-1 (insulin receptor substrate 1). Los IRS son moléculas que contienen múltiples residuos de tirosina y regiones reguladoras a base de sitios de serina y treonina (Ser/Tre), todos capaces de fosforilarse. Existen cuatro tipos IRS, de los cuales el 1 y 2 comparten un 80% de homología estructural, una de las cuales está representada por la región PTB (unión a Tir-fosfato), que sirve de unión al patrón NPXY (Asparagina-Prolina-XTirosina) de la región juxtamembrana del RI. La molécula de IRS contiene también varios sitios con residuos de tisrosina (Tir), que funcionan como anclaje para otras proteínas, con regiones tipo Homólogo a Src-2 (SH2). Ejemplo de esto es la región p58α de la fosfatidilinositol-3-cinasa (PI3k), proteína de unión al receptor de crecimiento 2 (Grb2), entre otras, las cuales finalmente favorecen las funciones metabólicas y de crecimiento celular dependientes de la insulina. Los eventos de señalización postreceptor involucran dos vías principales: 1) La vía de la PI3k6. 2) La proteincinasa activada por mitógenos (MAPK)7 (Ver Figura 1).

La Vía de la PI3k
La vía de la PI3k se inicia con la asociación de esta enzima con el IRS-1 en la región SH2 del complejo p58α/p110 de la PI3k, lo que da como resultado la activación de esta enzima (PI3k) y la producción de 3,4,5-fosfatidilinositol (PIP3). El PIP3 activa a la cinasa dependiente de PI3k (PDK-1) mediante su unión con la región PH de dicha cinasa, la cual a su vez fosforila a la enzima Akt (una cinasa Ser/Tre tipo B). Se ha implicado a la Akt en la traslocación de los GLUT4 y en la estimulación de la síntesis de glucógeno mediante la modificación covalente por fosforilación de la glucógeno sintetasa cinasa 3, cuyo resultado final es su inactivación, por lo que la glucógeno sintetasa queda libre para iniciar la producción de glucógeno. Además, por intermedio de esta vía se incrementa la sensibilidad de los miofilamentos al Ca++, se estimula la traslocación de la bomba de Na+ y los canales de K+ a la membrana y aumenta la síntesis de óxido nítrico (ON) por la inducción de la ON sintetasa9.
Vía de las mapcinasas (MAPK)
La vía de las MAPK, también denominadas vía de las ERK, constituye una de las vías de la activación final de las proteincinasas activadas por mitógenos llamadas ERK (extracellular-signal receptor kinase) tipo 1 ó 2. Estas moléculas se encargan de la modificación de la expresión de ciertos genes (c-fos, Elk-1), implicados en varios procesos biológicos, como el crecimiento y la diferenciación celular. La cascada se inicia con la unión del IRS-1 al dominio SH2 de la proteína Grb2, que se encuentra unida previamente al SOS (Son of Sevenless), una pequeña proteína de intercambio de nucleótidos que cataliza el intercambio de guanosindifosfato (GDP) por guanosintrifosfato (GTP) en la proteína Ras. Esta proteína es una GTPasa que se encuentra adosada a la cara interna de la membrana plasmática, unida en su extremo amino a Raf (Ras activating factor), favoreciendo la fosforilación de ésta última en sitios Ser/Tre, con activación de su función de cinasa (Raf-1). La molécula Raf-1 estimula a la MAPKcinasa, una enzima dual también conocida como MAPKcinasa/ERKcinasa por la fosforilación de dos sitios de serina reguladores. Estas enzimas tienen como función la activación de las ERK mediante la fosforilación de residuos de Tir y Tre. La regulación de ésta vía es probablemente realizada por la propia insulina, que estimula el funcionamiento de la MAPKfosfatasa (MKP-1)8, la cual desfosforila a la MAPK, deteniendo la cascada.
Otras Vías
La cara interna de la membrana celular está en contacto con mallas de fibras derivadas del citoesqueleto, que se ensamblan como puntos de adhesión focal (PAF), los cuales además de controlar la forma celular son capaces de transmitir señales extracelulares hacia el citosol. La regulación de éstos PAF es relativamente sencilla, mediante dos proteínas principales la Paxilina y la FAK (cinasa de adhesión focal), que se activan cuando son fosforiladas por cascadas de receptores con propiedad de tirosincinasa. Al ser estimuladas ocurre un reordenamiento del citoesqueleto, que propicia el ensamblaje de los PAK, generando las fibras de estrés. La insulina es capaz de producir ruptura de las fibras de estrés mediante la desfosforilación de FAK y paxilina, por la unión del IRS-1 con la proteína Csk (cinasa de la porción carboxilo terminal de Src)10.
Bases moleculares y celulares de la insulinorresistencia
La IR es un fenómeno que se observa en la diabetes mellitus tipo 2 (DM2), obesidad, síndrome de ovario poliquístico, infección crónica y el síndrome metabólico1. Para explicar el origen de la insulinorresistencia se han postulados varias teorías, entre ellas tenemos:
Obesidad y Ácidos Grasos Libres
La obesidad es una enfermedad que se ha convertido en un problema de salud pública11 tanto por su prevalencia como por su elevada relación con muchas enfermedades crónicas degenerativas (cáncer, DM2, hipertensión y aterosclerosis y sus complicaciones). Los estudios observaciones y clínicos que correlacionan estos aspectos12,15 han demostrado que la obesidad androide se asocia con un flujo elevado de ácidos grasos libres (AGL) en el lecho esplácnico (portal), asociado a la disminución de la inhibición de la lipólisis en el tejido adiposo dependiente de la insulina. Se conoce que los AGL contribuyen tanto a la aparición como a la progresión de la IR e hiperinsulinemia en pacientes obesos16,17, lo cual se produce por varios mecanismos: disminución de la captación y utilización de glucosa, inhibición del ciclo de Krebs y alteración en el patrón de secreción de insulina.
En 1963, Randle y colaboradoes18 propusieron el ciclo glucosa-ácidos grasos para explicar la relación inversa entre la sensibilidad a la insulina y el nivel de AGL séricos en ayuno, postulando que los AGL compiten con la glucosa como substrato energético en el músculo estriado (Ver Figura 2). Recientemente esta teoría ha sido cuestionada19,20, presentándose hipótesis alternativas como la postulada por Shulman y colaboradores21, en la cual el aumento de AGL conduce a un incremento de ciertos metabolitos intracelulares, como el diacilglicerol, Acil-Coa y ceramidas, que son capaces de activar Ser/Trecinasas (proteinacinasa Cθ), que fosforila en sitios de Ser/Tre al IRS 1 y 2, lo que reduce la habilidad del receptor para iniciar la vía de la PI3k y de esta forma la disminución del transporte de la glucosa (ver figura 3).


Sin embargo, el papel de los AGL no sólo involucra el metabolismo oxidativo de la glucosa, sino también la modificación del patrón de secreción de la insulina. El equipo de Stein22 analizó el papel de los ácidos grasos saturados e insaturados de cadena larga sobre el patrón de secreción de insulina durante una administración endovenosa rápida de glucosa, concluyendo que el papel insulinotrópico de los AGL depende de la longitud de la cadena (positivamente) y del grado de insaturación (negativamente).
2. Factor de Necrosis Tumoral-α
El factor de necrosis tumoral α (TNF-α) es una proteína de 26 kDa, sintetizada por fagocitos, adipocitos y por el músculo cardíaco y esquelético, en pequeñas cantidades. Esta hormona tiene varias células blanco, formando así parte de los mecanismos de defensa inmunológica, del proceso inflamatorio, caquexia y de IR6,23. Como es producida abundantemente por los adipocitos, los niveles de TNF-α se correlacionan positivamente con el grado de obesidad y la concentración de insulina plasmática, disminuyendo cuando mejora la sensibilidad a la insulina6. El TNF-α αtiene 2 receptores (TNFR-1 y 2) pertenecientes a la superfamilia de receptores para citocinas24 que incluye el factor de crecimiento neuronal (NGF) y el antígeno de superficie CD40. Se ha sugerido que el TNF-α interfiere tanto la señalización insulínica como la síntesis de los transportadores de glucosa23,24 produciendo IR en los adipocitos, mediante la fosforilación de residuos Ser/Tre en el IRS-1, gracias a la activación de Ser/Treonincinasas (subtipos de proteínas cinasas C).
3. Leptina
La leptina es el producto del gen Ob/Ob sintetizadas por el tejido adiposo blanco. Dentro de su espectro de acciones fisiológicas25 está la de inhibir la secreción de insulina por intermedio de su receptor ObRb, localizado en la célula α pancreática. También son capaces de disminuir el gasto energético y la ingesta de alimentos mediante la estimulación de sus receptores en el hipotálamo. La leptina reduce tanto la secreción como la síntesis de insulina mediante a través de tres mecanismos: A) El complejo JAK/STAT3-5 que actúa a nivel nuclear. B) La activación de la fosfodiesterasa 3B que reduce la disponibilidad de AMPc. C) Por la apertura de canales de K+ATP-sensibles, que impide la secreción de la insulina.
En el hígado, la leptina disminuye la capacidad de la insulina para inhibir la fosfoenolpiruvato carboxicinasa26 (PEPCK), enzima clave en la gluconeogénesis. Muchos autores sugieren que la obesidad es un estado de resistencia a la leptina, en el cual se crea un círculo vicioso hiperleptinemia-hiperinsulinemia/leptinoresistencia-insulinoresistencia, que precede y luego acompaña a la DM2).
4. Glucosa y vía de las hexosaminas
Es bien sabido que la hiperglicemia ejerce efectos adversos sobre la señalización insulínica, y uno de los mecanismos (y quizá el principal) es la derivación de la glucosa hacia la vía de las hexosaminas (Ver Figura 4). La enzima clave de esta vía es la glutamina:fructosa-6-fosfato aminotransferasa (GFA), y su producto final el Uridin Difosfato N-acetil-glucosamina (UDP-GlcNAc) es protagonista de la O-glicosilasión de factores de transcripción en células β y adipocitos27. Estos productos estimulan la expresión y síntesis de leptina. Sin embargo, éste no es el único mecanismo ya que las reorganizaciones genéticas disminuyen la síntesis de las proteínas encargadas de la traslocación de los GLUT428 (superfamilia de transportadores SNARE). La glucosamina produce IR en el músculo estriado, cardíaco, hígado y tejido adiposo, así como hiperinsulinemia compensadora29. Finalmente, Vosseller y colaboradores30 demostraron que un aumento de la O-glicosilación de la UDP-GlcNAc (O-GlcNAc) disminuye la fosforilación de Akt en el residuo de Tre308, lo cual detiene el paso final de la vía de la PI3k.

Insulinorresistencia, hiperinsulinemia e hipertensión arterial En 1987 Reaven y Hoffman31 propusieron que la IR, y más específicamente, la hiperinsulinemia, podrían estar involucradas en el desarrollo de la hipertensión arterial. Esta correlación persiste, especialmente por la asociación de enfermedad hipertensiva esencial (EHE) con elevación de la insulina en ayunas y postprandial32, en comparación con sujetos normotensos sin importar su índice de masa corporal. Además, se ha observado que los modelos animales para hipertensión (ratas hipertensas Dahl33, la rata espontáneamente hipertensa34 y la rata obesa e hipertensa Zucker35) presentan IR e hiperinsulinemia. Se ha propuesto la intervención de varios mecanismos para explicar esta relación (Ver Figura 5):
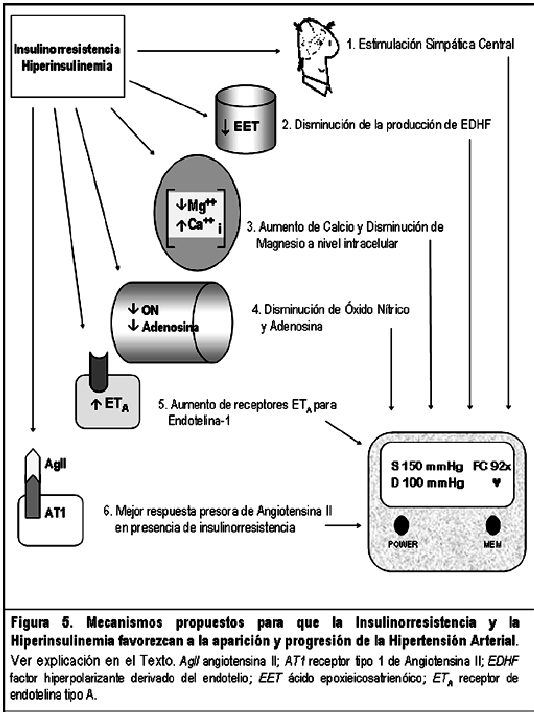
1. Hiperactividad simpática
Se conoce que la infusión de insulina e ingestión de carbohidratos estimula la actividad nerviosa simpática y que este efecto simpaticoexcitatorio es mediado centralmente36,38, ya que la insulina atraviesa la barrera hematoencefálica activando sus receptores en el hipotálamo medial. En ratas, cuando la insulina se administra intraventricularmente (no hay acción sistémica), solo se detecta aumento de la actividad simpática nerviosa39. El efecto simpaticoexcitatorio de la insulina se ha observado en sujetos insulino-resistentes no obesos, mientras que en los obesos pareciera estar abolida40, pudiendo este fenómeno ser producido por un efecto excitatorio sostenido41,42, que no obstante, se ha observado en sujetos obesos hiperinsulinémicos. Por otra parte, en estos últimos pacientes, infusiones de corta duración no producen el mismo efecto que en sujetos no obesos.
2. Defecto de Vasodilatación
Usando el modelo animal de ratas alimentadas con fructosa se ha documentado deterioro de la relajación dependiente del endotelio, definido como una respuesta disminuida a la acetilcolina y a la bradicinina. Este defecto en la relajación está relacionado con un factor que es independiente del ON y de la prostaciclina, sustancias ambas que inducen vasodilatación mediante la activación de canales de K+ dependientes de Ca++, y que se ha denominado factor hiperpolarizante derivado del endotelio43 (EDFH). La identidad de EDHF es desconocida pero se sospecha que es un metabolito del ácido araquidónico sintetizado por intermedio del citocromo P450 epoxigenasa, llamado ácido epoxieicosatrienoico44,45 (EET). Se ha demostrado en estudios con arterias mesentéricas de ratas insulino-resistentes que éstas no se relajan cuando son expuestas al EET46, de hecho se encontró un pequeña vasoconstricción en las mismas. Para explicar este fenómeno se ha propuesto una alteración en los mecanismos regulatorios del canal de K+ que impide su apertura, ya sea por disminución de la disponibilidad de Ca++ o debido a que las arterias de las ratas insulino-resistentes producen otro metabolito del ácido araquidónico, el ácido 20-hidroeicotretaenóico, el cual inactiva los canales de K+ dependiente de Ca++, reportado en otros estudios con resultados semejantes47,48.
3. Alteración del metabolismo de cationes bivalentes
Calcio: La insulina reduce el tono vascular mediante sus efectos en el metabolismo catiónico49, debido a: I) Atenúa el influjo de Ca++ en los miocitos lisos vasculares, disminuyendo los canales de Ca++ operados por voltaje y los mediados porreceptor. II) Aumenta la actividad de la ATPasa de Ca++ en la membrana plasmática y organelos intracelulares; III) Activa los canales de K+ dependientes de Ca++ por intermedio del ON. IV) Estimula la bomba ATPasa Na+/K+, tanto de forma transcripcional como posttranslacional. Así mismo, en estados de IR se observa aumento en la resistencia vascular y respuesta vasoconstrictora, lo cual se asocia a defectos en la corrientes de Ca++, especialmente si además hay disminución de la actividad de la bomba ATPasa Na+/K+. En las ratas espontáneamente hipertensas se ha observado una disminución de la subunidad α catalítica de la bomba ATPasa Na+/K+ junto a un aumento de Ca++ intracelular50.
Magnesio: El Mg++ es el segundo catión más abundante a nivel intracelular, formando parte de todas las reacciones de transferencia del ATP. En la IR se observa depleción de Mg++ libre intracelular, hipertensión esencial y aumento de la resistencia vascular periférica49. Los eritrocitos y miocitos lisos vasculares de estos sujetos presentan niveles elevados intracelulares de Ca++ con bajos niveles de Mg++ más pH alterado51. Igualmente, la deficiencia de Mg++ demostrada en estos estados puede contribuir a deprimir y hasta suprimir el metabolismo de la glucosa y la acción insulínica52,53, la cual depende de varias transferencias de ATP.
4. Oxido nítrico y adenosina
La insulina es capaz de activar la ON sintasa para producir ON54, el cual realiza su función vasodilatadora mediante la generación de GMPc y el secuestro de Ca++. Estudios recientes demuestran que la insulina puede inducir a la producción de adenosina55, la cual, mediante sus receptores A1 y A2, produce hiperpolarización de la célula muscular lisa por activación de los canales de K+, produciendo vasodilatación. Por lo tanto en estados de IR la acción de éstos dos poderosos vasodilatadores está comprometida, aumentando así la resistencia vascular periférica.
5. Endotelina-1
Las endotelinas son potentes vasoconstrictoras de 21 aa codificadas por 3 genes en diferentes tejidos del cuerpo. Endotelina 1 (ET-1) es la principal generada por el endotelio, actuando de forma paracrina y autocrina sobre los receptores ETA y ETB y sus efectos dependen del lecho vascular donde se encuentren. En miocitos lisos vasculares ambos inducen contracción, proliferación e hipertrofia celular, mientras que en el endotelio el ETB estimula la producción de ON y prostaciclina. En otros lechos, como en la circulación coronaria, el ET-1 actúa como vasoconstrictor56 debido a la ausencia de ETB en el mismo. Se ha establecido correlación entre los niveles de insulina, índice de masa corporal y ET-157, por lo tanto en hiperinsulinemia la ET-1 se encuentra elevada, además en estados de IR los receptores ETA se encuentran incrementados58, por ende la respuesta vasoconstrictora se encuentra aumentada en este cuadro clínico.
6. Sistema renina-angiotensina
Hay evidencia sobre las acciones hipertensinogénicas de la insulina y de la angiotensina II (AII). Brands y colaboradores59 reportaron que era necesario un sistema renina-angiotensina (RAS) intacto para que la insulina pudiese inducir hipertensión, ya que la AII tiene mejor respuesta presora en presencia de hiperinsulinemia. Así mismo, Rocchini y colaboradores60 reportaron que la insulina incrementa la respuesta de AII en células mesangiales murinas y que las ratas Zucker presentan mayor sensibilidad a la AII61. Existe un grupo especial de hipertensos denominado los No Moduladores caracterizados por un sistema RAS más sensible de lo normal con mayor sensibilidad a la sal; se cree que ésta modalidad tiene un fuerte componente genético62 ya que los no moduladores son homocigotos para Tre235 en el gen de angiotensina. Los no moduladores se caracterizan por la conjunción: IR/hiperinsulinemia/anormalidades lipídicas/historia familiar de infarto del miocardio/incremento en la actividad del transportador Na+/Li+ en el eritrocito63, por lo cual se ha postulado a la IR como marcador genético de dislipidemia familiar e hipertensión.
Insulinorresistencia, hiperinsulinemia y arritmias
La IR ocurre en tejidos usualmente insulino sensibles, como el hígado, tejido adiposo y músculo esquelético. Actualmente se sabe que el músculo cardíaco también exhibe IR en diferentes patologías como obesidad, hipertensión y enfermedad coronaria. Dentro de la esfera cardiovascular, se ha descubierto que la insulina es capaz de modular el flujo sanguíneo, el funcionamiento de la bomba cardíaca y ciertos canales iónicos, incluyendo el canal L de Ca++.
En el ventrículo de ratón se han caracterizado 4 tipos de canales para el potasio64: 1) Los canales de K+ rectificadores hacia dentro (IKI). 2) Los canales de K+ independientes de calcio con dirección hacia fuera (Ito). 3) Los canales rectificadores tardíos ultrarrápidos (IKur). 4) Los canales de K+ steadysteate (Iss) que actúa como una corriente sostenida del ión.
Shimoni y colaboradores65 han determinado cambios en los Iss en estados de hiperinsulinemia con una elevación crónica de las magnitudes de mismo, patrón que el metformin es capaz de revertir parcialmente. Este hallazgo es de particular importancia ya que la repolarización del cardiomiocito depende de un flujo iónico de Cl-, Ca++, Na+ y K+ adecuados. Según Shimoni, el prolongamiento de la repolarización mediado por defectos en la modulación insulínica del canal, produciría un alargamiento del intervalo QT con modificación del patrón de dispersión del QT67.
Insulinorresistencia, hiperinsulinemia y aterosclerosis
La aterosclerosis es un proceso que involucra varios sistemas y procesos celulares, como son: inmunológico, inflamatorio, crecimiento y proliferación celular, metabolismo de lípidos y trombosis y coagulación, todos los cuales puede conducir a la oclusión progresiva o súbita de la luz de la arteria que padece de la placa ateromatosa. Es un proceso que comienza desde la infancia (aunque muchos afirman desde la etapa prenatal) con la formación de la estría grasa y los sucesivos eventos (Ver Figura 6).

La base del proceso aterosclerótico es una injuria sostenida del endotelio gracias a un gran número de factores, entre los cuales se citan: cigarrillo, alcohol, hipercolesterolemia, complejos inmunes, estrés de la pared arterial por aumento de la presión arterial (sheer stress) y otros. El daño endotelial se manifiesta en un sin número de formas, sin embargo, lo más relevante es la pérdida de sus propiedades antiagregantes, la retención masiva de partículas lipídicas de tipo LDL en el subendotelio y el fallo de la maquinaria antioxidante. Con el tiempo, las LDL son oxidadas por radicales libres producidos por el propio endotelio (LDL-ox). Las LDL-ox son capaces de inactivar a la ON sintasa, inducir la quimiotaxis de los polimorfonucleares y de las células musculares lisas de la capa media, y producción de matriz extracelular. Por su parte, los macrófagos que se encuentran en la subíntima tienen en su membrana un receptor barrendero denominado Scavenger, capaz de detectar y fagocitar a las LDL-ox, las cuales se van acumulando hasta que el macrógafo se encuentra lleno de éstas (células espumosas) y muere por explosión. Al morir, el macrófago libera LDL-ox a medio digerir y material oxidativo derivado de su lisosomas, agravando el escenario oxidativo subendotelial. A continuación, comienzan a depositarse detrito celular, LDL-ox, células inmunológicas y matriz extracelular en el lugar de la injuria, formándose la placa ateromatosa.
Según la American Heart Association on Vascular Lesions, las placas pueden dividirse en 6 tipos69, donde la estría grasa es el tipo III, las placas vulnerables son tipo IV-Va, y la placa complicada es tipo VI. De particular interés son los tipos IVVa, ya que ligeros cambios en su microambiente causarían lo que se denomina accidente de la placa (cambio de geometría más trombosis aguda) transformándolos en tipo VI. Este tipo de fenómeno es el primero de una serie de eventos trombóticos que llevarán al infarto del miocardio y a enfermedades cerebrovasculares de tipo isquémico-trombótico. Entre los factores implicados70 en estos accidentes tenemos hipercolesterolemia, Lp(a), aumento de fibrinógeno y del inhibidor del activador de plasminógeno-1 (PAI-1).
Se ha sugerido que niveles elevados de insulina (o reflejados en elevados niveles de IR) se relacionan con el desarrollo de aterosclerosis71, tanto por efecto directo sobre la pared arterial, como indirectamente a través de sus efectos en lípidos y presión arterial. A continuación se presentan los principales efectos de la IR e hiperinsulinemia para el desarrollo acelerado de la aterosclerosis:
1. Dislipidemia
La insulina es la encargada de inhibir la maquinaria lipolítica en el tejido adiposo, lo cual indirectamente inhibe la secreción de VLDL/TAG a partir del hígado. Sin embargo, en estados de hiperinsulinemia aunque al principio existe la supresión, luego aparece una estimulación de la secreción de estas partículas lipoprotéicas. Se postula que este fenómeno de estimulación hiperinsulinémica se debe a la presencia de ácidos grasos no esterificados (NEFA) los cuales provienen de un tejido adiposo IR, capaces de estimular la gluconeogénesis, favorecer la secreción de VLDL a partir del hígado, favorecer a la acumulación de TAG en la célula β, lo cual produce una secreción inadecuada de insulina y por último, iniciar la apoptosis de la misma72.
La IR se caracteriza por un patrón lipídico73 generalmente constante: niveles elevados de VLDL, LDL de pequeño tamaño y Lp(a) más una disminución de las HDL. El incremento de triacilglicéridos (TAG) en forma de VLDL puede entrar a la pared vascular acumulándose en las placas ateroscleróticas, además estas lipoproteínas pueden recibir ésteres de colesterol (EC) a través de la CETP (proteína de transferencia de esteres de colesterol), capaz entonces de transportar más colesterol a la pared vascular. Bajos niveles de HDL y apo A-I reflejan la incapacidad de éstas para ejercer sus funciones antiaterogénicas, entre ellas, recolectar el colesterol de las paredes vasculares hacia el hígado y actuar como antioxidante. Cuando la balanza se encuentra a favor de los VLDL, las HDL se empobrecen de EC por lo que su receptor scavenger B1 en el hígado, encargado de llevar el núcleo lipídico hacia el hepatocito sin necesidad de endocitosis ni degradación de la partícula completa, disminuye su capacidad de captación. Las partículas de LDL pequeñas y densas son más aterogénicas ya que son más propensas a oxidación y son más fáciles de adherirse a las células endoteliales de la pared, por lo tanto aceleran el proceso aterosclerótico. La dieta CAP (cronobiológica, antioxidante y polarizante) ha demostrado su utilidad al permitir que se complete el ciclo de eliminación hepático, que sucede, coincidentemente en el mismo horario del ritmo circadiano, el de mayor susceptibilidad cardíaca, de 4:00 AM a 12:00 M.
2. Hiperhomocisteinemia
La homocisteína es un aa que deriva de la metionina, la cual puede ser transformada a su aa de origen por acción de la metioninsintetasa74, pero ésta enzima depende de la producción de 5-metil tetrahidrofolato mediante la 5,10-metilentetrahidrofolato reductasa (MTHFR). El nivel de insulina influye en el metabolismo de la homocisteína75, probablemente por estimulación de la MTHFR ó de la cistation α-reductasa. Por ende, en estados de IR, la activación dada por la insulina se verá aplacada, favoreciendo a la hiperhomocisteinemia.
La homocisteína es capaz de producir disfunción endotelial, por inhibición de la relajación inducida por el endotelio76, que depende de la producción de ON, relacionada a su vez con la producción de radical anión superóxido (O2-), el cual parece provenir de la propia molécula de homocisteína. Se sabe que el O2- se une al ON formando peroxinitrito, que además de favorecer a la disfunción endotelial es capaz de inducir apoptosis76.
3. Perfil de Coagulación
Se han observado ciertos disturbios en el sistema de la coagulación asociados a IR e hiperinsulinemia, entre ellos tenemos:
PAI-1: El PAI-1 es sintetizado por hepatocitos, adipocitos y células endoteliales. La insulina estimula la síntesis de ésta proteína mediante una correlación interesante entre la vía de la PI3k y la MAPK. En efecto, al unirse la insulina con su receptor fosforila a IRS-1, éste se une a PI3k, la cual activa a varios subtipos de proteincinasa C (ζ, η, λ, ε), todas capaces de fosforilar y activar a la MEK-1, que a su vez induce la actividad de ERK2. La ERK2 se une a la región Sp177, complejo nuclear de factores de transcripción, estimulando así la síntesis de ARNm de PAI-1. Abbasi y colaboradores78 determinaron de forma inequívoca que los niveles del PAI-1 eran más elevados en mujeres insulinorresistentes, sin considerar edad, status menopáusico, terapia de reemplazo hormonal, obesidad (IMC), distribución del tejido adiposo ni presión arterial. Gracias a éste y otros estudios78,79, se considera que el grado de hiperinsulinemia se correlaciona con los niveles séricos de PAI-1.
El Sistema Fibrinogénico: Dentro del cuadro clínico de la IR se ha determinado que existe un estado de hipercoagulabilidad, caracterizado por niveles elevados de fibrinógeno80 junto a una deficiencia de los factores C y S de la antitrombina III49, encargados de inhibir la formación del coágulo.
4. Proteína C reactiva
La proteína C reactiva (PCR) es una proteína de fase aguda la cual se une a vesículas que contienen isofosfatidilcolina y lipoproteínas. La PCR aparece en la enfermedad cardiovascular con daño a tejido miocárdico (isquemia y necrosis miocárdica) o sin él81, indicando la extensión y gravedad de la aterosclerosis. Se ha determinado que la PCR se asocia a IMC, TAG, HDL (inversamente) y presión arterial82. Aunque se ha establecido una relación entre los niveles de ésta proteína e IR/hiperinsulinemia, no se conocen muy bien los mecanismos. Sin embargo, se ha postulado varias hipótesis82: I) La inflamación crónica es el factor detonante del síndrome IR/hiperinsulinemia, lo que eventualmente llevará a diabetes mellitus 2. De acuerdo a ésta teoría la sobrenutrición produce una hipersecreción de citocinas (TNF-α IL-1 y IL-6) que interfieren el metabolismo intermediario y son capaces de estimular la síntesis de PCR. II) La disminución de la sensibilidad a la insulina aumenta la síntesis de PCR por falta de de inhibición de la síntesis de proteínas de fase aguda. III) La inflamación tiene relación con los TAG y con el tejido adiposo corporal, porque la producción de TNF-α será mayor o menor de acuerdo al tipo de ácidos grasos que compongan los TAG.
5. Disfunción plaquetaria
La insulina sensibiliza las plaquetas a la acción antiagregante del ON y de la prostaciclina, a la vez que disminuye sus propiedades proagregantes. Se sabe que en éstas células existen receptores de insulina funcionantes, los cuales regulan en alta los receptores para prostaciclina, al mismo tiempo que regulan en baja los α-adrenérgicos. Además la insulina impide la unión de las plaquetas al colágeno in vivo, efecto que se ve abolido en estado de IR83. Por lo tanto, las plaquetas en este ambiente presentan incremento del número de receptores para agonistas y para proteínas de adhesión, disminución de la fluidez de la membrana plasmática, alteración del recambio de fosfolípidos de membrana y respuestas defectuosas ante antagonistas, características que favorecen a los fenómenos trombóticos oclusivos a nivel de las placas ateromatosas.
Insulinorresistencia, hiperinsulinemia e isquemia miocárdica
El metabolismo normal del miocardio involucra la oxidación de AGL durante el ayuno, y oxidación de la glucosa durante el estado postprandial. El primer paso del metabolismo de la glucosa en el cardiomiocito es su paso a través del sarcolema, el cual realiza mediante transportadores de glucosa tipo GLUT 1 y 4. El GLUT1 es el responsable del transporte basal del la glucosa en los cardiomiocitos, para luego ser traslocados velozmente al sarcolema en respuesta a la isquemia. Por otro lado, el GLUT4 se trastoca a la membrana desde su pool intracelular gracias al estímulo de la insulina, mientras que durante la isquemia miocárdica84 lo hace por otros mecanismos activados por la hipoxia.
La hibernación86,87 es un proceso adaptativo que aparece cuando existe un desequilibrio entre la función contráctil miocárdica y el flujo sanguíneo, alcanzando un estado metabólico que previene la necrosis. Durante la isquemia el tejido cardíaco se caracteriza por:
Disminución tanto en la producción como en el almacenamiento de ATP.
Incremento de glucógeno
Acidificación del citosol mediante el lactato producido por la glucólisis anaerobia
Liberación de Ca++ por parte del retículo sarcoplásmico asociada a una mayor activación de los canales de K+ dependientes de ATP por estimulación de la adenosina y el aumento de lactato.
Acumulo de Fosfato (Pi) el cual reduce la función contráctil del cardiomiocito mediante su unión a proteínas contráctiles e inhibición de la ATPasa miofibrilar.
Activación de enzimas lisosómicas y producción de radicales libres.
Reducción de más de 50% de la función contráctil del miocardio mientras se lleva a cabo un cambio en el metabolismo energético a base de glucosa, el cual consume menos energía y produce menos cantidad de especies reactivas de oxígeno.
En estados de normoxia existe la degradación permanente dependiente de la ubiquitina del factor inducible por hipoxia-1α (HIF-1α)87. Sin embargo, durante la hipoxia esta degradación es limitada, dejando libre a esta proteína, la cual forma un heterodímero con el llamado traslocador nuclear arilhidrocarbono (ARNT). Este complejo HIF-1α/ARNT es el encargado de estimular la síntesis y traslocación de GLUT1, GLUT4, enzimas de la vía glicolítica y factor de crecimiento endotelial vascular (VEGF) cuando el lecho isquémico se recupera. Todas estas inducciones van a permitir la regeneración de vasos sanguíneos que ayudarán a una revascularización efectiva y a poner en marcha la maquinaria glicolítica mientras se recupera la función contráctil.
Un mecanismo modulador del complejo HIF1α/ARNT es producido por la insulina. Zelzer y colaboradores88 comprobaron que la insulina es capaz de mediar parte de sus efectos angiogénicos mediante la activación del complejo HIF1α/ARNT, aunque no se ha sido esclarecido su mecanismo. Por lo tanto, el éxito de la revascularización total y la supervivencia del cardiomiocito dependen de una cascada insulínica íntegra.
Iozzo y colaboradores89 demostraron que en la hiperinsulinemia hay una respuesta disminuida con respecto a la toma de glucosa, asociado a una disminución de ARNm para GLUT1 y GLUT4, por lo tanto se puede inferir que en los primeros minutos de hipoxia, la traslocación inicial de las vesículas con transportadores presintetizados se ve comprometida, reduciendo las posibilidades de supervivencia del miocito isquémico. Además, el estimulo angiogénico necesario para la formación de nuevos vasos que permitan restituir un flujo acorde a las demandas energéticas del músculo se ve disminuido porque la cascada insulínica no podrá estimular la formación y estabilización del complejo HIF1α/ARNT. La radical importancia de un sistema insulínico intacto y mecanismos que permitan un aporte de glucosa se evidencian en los efectos beneficiosos de las perfusiones de glucosa-insulina-potasio (GIK)90 en pacientes con infarto del miocardio, en el cual se observa una reducción del tamaño del infarto y mejoría de la función ventricular.
Insulinorresistencia, hiperinsulinemia y otras patologías cardiovasculares
1. Enfermedad Cerebrovascular
Las enfermedades cerebrovasculares (ECV) son una de las principales causas de déficit neurológico y muerte en los últimos años a pesar de un gran avance en las técnicas de manejo agudo del cuadro. Las ECV puede ser de tipo isquémico (trombótico in situ o embólico cardíaco, carotídeo o aórtico) o hemorrágico. Los arteriales se originan en placas ateromatosas, mientras que los de origen cardíaco se relacionan con la formación de trombos murales en las paredes atriales o ventriculares (izquierdas) de sujetos que padecen trastornos cardíacos como arritmias, cardiomiopatías o infarto del miocardio. El infarto lacunar (silencioso) es un subtipo de ECV isquémico el cual se caracteriza por lesiones pequeñas, menores de 5mm, que suceden en las arteriolas que irrigan los ganglios basales, tallo cerebral, cerebelo y materia blanca profunda. Este tipo de infarto se asocia a hipertensión de larga data, por degeneración hialínica y necrosis fibrilar de sus arteriolas. Entre los principales factores de riesgo para ECV91 están: EHE, hiperlipidemia, DM2, hábito tabáquico, enfermedad cardíaca, SIDA, abuso de drogas, alcoholismo e historia familiar de ECV.
Si tomamos en cuenta los principales factores de riesgo mencionados anteriormente, la IR y la hiperinsulinemia prolongan y dificultan el tratamiento de dichos factores, por lo tanto, un estado insulinorresistente incrementaría indirectamente las posibilidades para ECV. Shinozaki y colaboradores92 analizaron 34 pacientes con ECV de tipo trombótico, cardioembólico y lacunar, determinando que la hiperinsulinemia se correlacionaba con los ECV isquémicos y no con los lacunares. Además, establecieron que la contribución de la IR y la hiperinsulinemia en la aparición de estos cuadros era la de iniciar y mantener niveles elevados de TAG, aumento de la presión arterial y disminución de la HDL. Por su parte, Erdos y colaboradores93 examinaron el sistema de canales iónicos de las arterias de mediano calibre a nivel cerebral y establecieron que la IR influye en el metabolismo de los canales de K+ATP y K+Ca++, por lo que la regulación del tono vascular se ve afectado. Por tanto, no es de sorprender que dentro de unos años, la IR y su hiperinsulinemia compensatoria sean factor de riesgo para ECV de tipo hemorrágico.
2. Hipertrofia ventricular izquierda (HVI)
El remodelamiento cardíaco94 ocurre en respuesta a un reordenamiento de las estructuras ya presentes en el miocardio del ventrículo izquierdo, término que se ha relegado para hablar de enfermedad cardiovascular (ECV). El reordenamiento (mejor que remodelamiento) abarca tanto el miocardio como los vasos sanguíneos, estrés mecánico, isquemia, hormonas, péptidos vasoactivos, aumento de la precarga y cicatrices miocárdicas. La hipertrofia ventricular izquierda95 también forma parte del espectro de ese reordenamiento cardíaco, caracterizado por una hipertrofia del cardiomiocito (reorganización sarcomérica) e hiperplasia de fibroblastos y células endoteliales. El aumento del número de fibroblastos produce fibrosis debido a un incremento en la producción de colágeno (sobre todo el Tipo I), lo que conduce a un aumento de la rigidez miocárdica (falsa hipertrofia), heterogeneidad eléctrica (dispersión del QT) y actividad arritmogénica, especialmente por ser una de las formas más comunes del mecanismo de la reentrada.
Se ha relacionado la IR e hiperinsulinemia como factores asociados a la hipertrofia ventricular. McNulty y colaboradores96 demostraron que la hiperinsulinemia se correspondía con una disminución del metabolismo proteico miocárdico (medido por el balance de la fenilalanina), que conduce a un enlentecimiento del recambio de las fibras colágenas. Las colagenasas destinadas a degradar el colágeno son activadas principalmente por la plasmina, cuya acción depende del activador de plasminógeno tisular. Por otra parte, en IR la síntesis del PAI-1 está aumentada, que compromete el trabajo efectivo de las colagenasas. Por último, Abel y colaboradores97 utilizando modelos animales knock-out y heterocigotos para el gen de GLUT4 concluyeron que la hiperinsulinemia y el defecto de la bomba cardíaca inducen y agravan la hipertrofia ventricular de éstos animales. Hasta el presente muchos autores manifestaron predilección por la ecocardiografía para establecer el diagnóstico de HVI.
Referencias
1. Williams, B. Insulin resistance: the shape of things to come. Lancet 1994; 344:521-524. [ Links ]
2. Himsworth, H. Diabetes Mellitus: its differentiation into insulin-sensitive and insulin-insensitive types. Lancet 1936; 1:127-130. [ Links ]
3. Luo R, et al. Quaternary structure of the Insulin-Insulin Receptor Complex. Science 1999;283:1077-1080. [ Links ]
4. White, M. The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Rescent Prog Horm Res 1998; 53:119-138. [ Links ]
5. Cheatham B, Kahn C. Insulin action and the insulin signaling network. Endocr Rev 1995; 16:117-142. [ Links ]
6. Le Roith, D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care 2001; 24:588-597. [ Links ]
7. Pearson G, et al. Mitogen Activated Proteins (MAP) kinase pathways: regulation and physiological functions. Endocrin Rev 2001;22(2):153-183. [ Links ]
8. Kusari A, et al. Insulin-Induced Mitogen-Activated Protein (MAP) Kinase Phosphatase-1 (MKP-1) Attenuates Insulin-Stimulated MAP Kinase Activity: A Mechanism for the Feedback Inhibition of Insulin Signaling. Mol Endocrinol 1997;11(10):1532-1543. [ Links ]
9. Sadagurski M, Weingarten G. Insulin Receptor Substrate 2 Plays Diverse Cell-specific Roles in the Regulation of Glucose Transport. J Biol Chem 2005; 280:14536-14544. [ Links ]
10. Wang Q, Bilan P, Klip A. Opposite effects of insulin on focal adhesion proteins in 3T3-L1 adipocytes and in cells overexpressing the insulin receptor. Mol Biol Cell 1998;9(11):3057-3069. [ Links ]
11. WHO Nutrition Data Banks Global Database on Obesity and Body Mass Index (BMI) in Adults. http://www.who.int/nut/db_bmi.htm [ Links ]
12. Wajchenberg, B L. Subcutaneos and Visceral Adipose Tissue: their relationship to the Metabolic Syndrome. Endocrine Reviews 2000;21(6):697-738. [ Links ]
13. Hill, J O., Peters, J C. Environmental Contributions to the Obesity Epidemic. Science 1998; 280(5368): 1371-1374. [ Links ]
14. Bjorntorp, P. The association between obesity, adipose tissue distribution and disease. Acta Med Scand Suppl. 1988; 723: 121-134. [ Links ]
15. Derpress, J P. Abdominal obesity an important component of insulin resistance syndrome. Nutrition 1999; 9:452-459. [ Links ]
16. Koyama, K., Chen, G., Lee, Y., Unger, R. Tissue Triglycerides, insulin resistance and insulin production: implications for hyperinsulinemia of obesity. Am J. Physiol Endrocrinol Metab 1997;273:E708-E713. [ Links ]
17. Kahn B B., Flier, J S. Obesity and Insulin Resistance. J Clin Invest 2000;106(4):473-481. [ Links ]
18. Koska J, Ortega E. The effect of insulin on net lipid oxidation predicts worsening of insulin resistance and development of type 2 diabetes mellitus. Am J Physiol Endocrinol Metab 2007; 293: E264- E269. [ Links ]
19. Roden M, et al. Mechanism of free fatty acids-induced insulin resistance in humans. J Clin Invest 1996; 97:2859-2865. [ Links ]
20. Griffin M, et al. Free fatty induced insulin resistance is associated with activity of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48:1270-1274. [ Links ]
21. Shulman, G. Cellular mechanism of insulin resistance. J Clin Invest 2000; 102(2):171-176. [ Links ]
22. Stein D, et al. The Insulinotropic Potency of Fatty Acids Is Influenced Profoundly by Their Chain Length and Degree of Saturation. J Clin Invest 1997; 100(2):398-403. [ Links ]
23. Hotamisligil G, Spiegelman B. Adipose expression of tumoral necrosis factor α: direct role in obesity-linked insulin resistance. Science 1993; 259:87-91. [ Links ]
24. Qi C, Pekala P. Tumor necrosis factor-α-induced insulin resistance in adipocytes. P.S.E.B.M 2000; 233:128-135. [ Links ]
25. Kieffer TJ., Habener, JF. The adipoinsular axis; effects of leptin on pancreatic α cell. Am J Physiol Endocrinol Metab. 2000;278:E1-E14.2000. [ Links ]
26. Taylor S, Barr V, Reitman M. Does leptin contribute to diabetes caused by obesity?. Science 1996; 274(5290):1151-0. [ Links ]
27. Emilsson V, et al. Hexosamines and Nutrient Excess Induce Leptin Production and Leptin Receptor Activation in Pancreatic Islets and Clonal ß-Cells. Endocrinology 2001;142(10):4414-4419. [ Links ]
28. Hebert L, et al. Overexpression of Glutamine: Fructose-6-Phosphate Aminotransferase in Transgenic Mice Leads to Insulin Resistance. J Clin Invest 1996;98(4):930-936. [ Links ]
29. Virkamäki A, et al. Activation of the Hexosamine Pathway by Glucosamine in Vivo Induces Insulin Resistance in Multiple Insulin Sensitive Tissues. Endocrinology 1997;138(6):2501-2507. [ Links ]
30. Vossller K, et al. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA 2002;99(8):5313-5318. [ Links ]
31. Reaven G, Hoffman B. A role for insulin in the a etiology and course of hypertension?. Lancet 1987;11:435-37. [ Links ]
32. Saad M., Rewers M, et al. Insulin Resistance and Hypertension. Hypertension. 2004;43:1324. [ Links ]
33. Kotchen TA, Zhang HY, Covelli M, Blehschmidt N. Insulin resistance and blood pressure in Dahl rats and in one-kidney, one-clip hypertensive rats. Am J Physiol 1991;261:E692–E697. [ Links ]
34. Reaven GM, Chang H. Relationship between blood pressure, plasma insulin and triglyceride concentration, and insulin action in SHR and WKY rats. Am J Hypertens. 1991;4:34–38. [ Links ]
35. Standley PR, Ram JL, Sowers JR. Insulin attenuation of vasopressin-induced calcium responses in arterial smooth muscle from Zucker rats. Endocrinology. 1993;133:1693–1699. [ Links ]
36. Hofman P. L., Regan F., Et all. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J. Clin. Invest., 2004; 114(5): 652 - 658. [ Links ]
37. Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 1999;34(part 2):724-728. [ Links ]
38. Margolis RU, Altszuler N. Insulin in the cerebrovascular fluid. Nature. 1967;215:1375–1376. [ Links ]
39. Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol. 1994;267:R1350–R1355. [ Links ]
40. Vollenweider P, Randin D, Tappy L, Jéquier E, Nicod P, Scherrer U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest. 1994;93:2365–2371. [ Links ]
41. Scherrer U, Randin D, Tappy L, Vollenweider P, Jéquier E, Nicod P. Body fat and sympathetic nerve activity in healthy subjects. Circulation. 1994;89:2634–2640. [ Links ]
42. Grassi G, Seravalle G, Cattaneo BM, Bolla GB, Lanfranchi A, Colombo M, Giannattasio C, Brunani A, Cavagnini F, Mancia G. Sympathetic activation in obese normotensive subjects. Hypertension. 1995;25(part 1):560–563. [ Links ]
43. Verma S, Bhanot S, Yao L, McNeill J. Defective endothelium-dependent relaxation in fructose-fed hypertensive rats. Am J Hypertens 1996;9:370-376. [ Links ]
44. Campbell W, et al. Identification os epoxyeicosatrienoic acid as endothelium derived hyperpolarizing factors. Circulation Research 1996; 78:415-423. [ Links ]
45. Capdevila J. H., Falck J. R., Harris R.C. Cytochrome P450 and arachidonic acid bioactivation: molecular and functional properties of the arachidonate monooxygenase. J.Lipid Res. 2000;41:163-181. [ Links ]
46. Miller A, et al. Epoxyeicosatrienoic acid-induced relaxation is impaired in insulin resistance. AJP-Heart Cir Physiol. 2001;281(4): H1524-H1531. [ Links ]
47. Katakam P, Ujhelyi M, Miller A. EDHF mediated relaxation is impaired in fructose fed rats. J Cardiovasc Pharmacol 1999;34:461-467. [ Links ]
48. Miller A, Hoenig M, Ujhelyi M. mechanism of impaired endothelial function associated with insulin resistance. J Cardiovasc Pharmacol 1998;3:125-134. [ Links ]
49. McFarlane S, Banerji M, Sowers J. Insulin resistance and cardiovascular disease. J Ckin Endocrin & Metab. 2001; 86(2):2713-2718. [ Links ]
50. Sowers J. Insulin and IGF in normal and pathological cardiovascular physiology. Hypertension 1997; 29:691-699. [ Links ]
51. Sowers JR. Effects of insulin and IGF-1 on vascular smooth muscle glucose and cation metabolism. Diabetes. 1996;45:47-51. [ Links ]
52. Chambers et al. Serum Magnesium and Type-2 Diabetes in African Americans and Hispanics: A New York Cohort. J. Am. Coll. Nutr. 2006;25:509-513. [ Links ]
53. Dominguez L, Sowers J, Barbagallo M, Resnick L. Magnesium responsiveness to insulin and IGF-1 in erythrocytes from normotensives and hypertensive subjects. J Clin Endocrin Metab 1998; 83(12):4402-4407. [ Links ]
54. Zeng G. Roles for insulin receptor, PI3-kinase, and Akt in insulinsignaling pathways related to production of nitric oxide un human vascular endothelial cells. Circulation 2000;101:1539-1545. [ Links ]
55. McKay M, Hester R. Role of nitric oxide, adenosine, and ATP sensitive potassium channels in insulin-induced vasodilation. Hypertension. 1996;28:202-208. [ Links ]
56. Schiffrin E. Role of Endothelin-1 in hypertension. Hypertension, 1999; 34(part 2):876-881. [ Links ]
57. Irving R, et al. Activation of the endothelin system in insulin resistance. Q J Med. 2001;94:321-326. [ Links ]
58. Katakam P, Pollock J, Pollock D, Ujhelyi M, Miller A. Enhanced endothelin-1 response and receptor expression in small mesenteric arteries of insulin-resistance rats. AJP - Heart Cir Physiol. 2001; 280(2):H522-H527. [ Links ]
59. Brands M, et al. Insulin-induced hypertension in rats depends on an intact rennin-angiotensin system. Hypertension. 1997;29:1014-1019. [ Links ]
60. Rocchini AP, Moorehead C, DeRemer S, Goodfriend TL, Ball DL. Hyperinsulinemia and the aldosterone and pressor responses to angiotensin II. Hypertension. 1990;15:861-866. [ Links ]
61. Zemel MB, Peuler JD, Sowers JR. Hypertension in insulin-resistant Zucker obese rats is independent of sympathetic neural support. Am J Physiol. 1992;262:E368-E371. [ Links ]
62. Ferri C, et al. Relationship between insulin resistance and nonmodulating hypertension. Linkage of metabolic anormalities and cardiovascular risk. Diabetes 1999;48:1623-1630. [ Links ]
63. Doria A, Fiotetto P, Avogaro A. Insulin resistance is associated with high sodium-lithium countertransport in esscential hypertension. Am J Physiol 1991;261:E684-E691. [ Links ]
64. Xu H, Gui W, Nerbonne J. Four kinetically distinct depolarizationactivated K+ currents in adult mouse ventricular myocytes. J Gen Physiol 1999;113:661-677. [ Links ]
65. Shimoni Y, Severson D, Ewart H. Insulin resistance and the modulation of cardiac K+ currents. AJP- Heart Cir Physiol. 2000; 279(2):H639-H649. [ Links ]
66. Bermúdez-Arias, F. Electrocardiografía diagnóstica. Editorial Mc-Graw-Hill Interamericana de Venezuela. Caracas 2000, pp 64-36. [ Links ]
67. Watanabe T, Ashikaga T, et al. Association of insulin with QTc dispersion. Lancet 1997;350:1821-1822. [ Links ]
68. Viskin S. Long QT syndromes and torsade de pointes. Lancet 1999;354 :1625. [ Links ]
69. Stary H, Chandler A, Sisismore R, et al. A definition of advanced type of atherosclerotic lesions and a histological classification of atherosclerosis. Classification of atherosclerosis. A report from the Committee on Vascular Lesion of the Council on Atherosclerosis. AHA. Circulation 1995;92:1355-1374. [ Links ]
70. Fuster V, Fayad Z, Badimon J. Acute coronary syndromes: biology. Lancet 1999;353(suppl II):5-9. [ Links ]
71. Howard G, et al. Insulin Sensitivity and atherosclerosis. The Insulin Resistance Atherosclerosis Study. Circulation. 1996;93:1809-1817. [ Links ]
72. Zammit V, Waterman I, Topping D, McKay G. Insulin stimulating of hepatic triacylglycerol secretion and the etiology of insulin resistance. J Nutr 2001;131:2074-2077. [ Links ]
73. Ginsberg H. Insulin resistance and cardiovascular disease. J Clin Invest. 2000; 106(4):453-458. [ Links ]
74. Durand P, Prost M, Loreau N, et al. Impared homocysteine metabolism and atherotrombotic disease. Lab Invest 2001;81:645-672. [ Links ]
75. Meigs J, et al. Fasting plasma homocysteine levels in the insulin resistance syndrome. The Framingham Offspring Study. Diabetes Care. 2001; 24:1403-1410. [ Links ]
76. Lang D, et al. Homocystein-induced inhibition of endothelium-dependent relaxation in rabbit aorta. Art Thromb Vasc Biol 2000;20:422. [ Links ]
77. Banfi C, et al. Transcriptional regulation of plasminogen activator inhibitor type 1 gen by insulin. Diabetes 2001;50:1522-1530. [ Links ]
78. Abbasi F, et al. Comparison of Plasminogen Activator Inhibitor-1 concentration in insulin-resistant versus insulin-sensitive healthy women. Arterios Thromb Vasc Biol. 1999;19:2818. [ Links ]
79. Juhan-Vague I, Alessi M. PAI-1, obesity, insulin resistance and cardiovascular events. Thromb Haemost 1997;78:656-660. [ Links ]
80. Válek J, et al. Increased fibrinogen levels in the offspring of hypertensive men. Relation with Hyperinsulinemia and the metabolic syndrome. Art Thromb Vasc Biol 1995;15:2229-2233. [ Links ]
81. Lagrand W, et al. C-Reactive protein as a cardiovascular factor. More than an epiphenomenon?. Circulation 1999;100:96-102. [ Links ]
82. Festa A, et al. Chronic subclinical inflammation as part of the insulin resistance syndrome. The Insulin Resistance Atherosclerosis Study. Circulation 2000;102:42. [ Links ]
83. Westerbacka J, et al. Inhibition of platelet-collagen interaction. An in vivo action of insulin abolished by insulin resistance in obesity. Art Thromb Vasc Biol 2002;22:167. [ Links ]
84. Russell R, et al. Additive effects of hiperinsulinemia and ischemia on myocardial GLUT1 and GLUT4 translocation in vivo. Circulation 1999;98:2180-2186. [ Links ]
85. Heusch G. Hibernating myocardium. Physiol rev 1998;78(4):1055-1085. [ Links ]
86. Gamici P, et al. Pathophysiological mechanisms of chronica reversible left ventricular dysfunction due to coronary artery disease (hibernating myocardium). Circulation 1997;96:3205-3214. [ Links ]
87. transportadores de glucose. [ Links ]
88. Zelzer E, Levy Y, et al. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO 1998;17:5085-5094. [ Links ]
89. Iozzo P, et al. Independent association of type 2 diabetes and coronary artery disease with myocardial insulin resistance. Diabetes. 2002;51:3020-3024. [ Links ]
90. Diaz R, Paolasso E, et al. Metabolic modulation of acute myocardial infarction. The ECLA Glucose-Insulin-Potassium pilot trial. Circulation 1998;98:2227-2234. [ Links ]
91. Sacco R. Identifying patient populations at high risk for stroke. Neurology 1998;51(Suppl 3):S27. [ Links ]
92. Shinozaki K, et al. Role of insulin resistance with compensatory hiperinsulinemia in ischemic stroke. Stroke 1997;27:37-43. [ Links ]
93. Erdos B, et al. Alterations in calcium-activated and ATP-dependent potassium channel function in cerebral arteries of insulin resistant rats. AJP-Heart Cir Physiol 2002;10:1152. [ Links ]
94. Swynghedanw, B. Molecular mechanisms of myocardial remodeling. Physiol rev 1999;79(1):215-262. [ Links ]
95. Weber T, Brilla C, Janicki J. Myocardial fibrosis : its functional significance and regulating factors. Cardiovasc Res 1993;27:341-348. [ Links ]
96. McNulty P, et al. Hyperinsulinemia inhibits myocardial protein degradation in patients with cardiovascular disease and insulin resistance. Circulation 1995;92:2151-2156. [ Links ]
97. Abel ED, Kaulbach H, et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 1999;104:1703-714. [ Links ]
