











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
INTRODUCTION
Breast cancer is the most frequently diagnosed cancer and the leading cause of cancer-related death in women worldwide, with an estimated 2.3 million new cases per year and approximately more than 685,000 deaths by 2020 1. Among treatments against this disease is the use of tamoxifen (Nolvadex ®) for cancer cells expressing estrogen receptors or trastuzumab (Herceptin®) for HER-2+ mammary tumors 2. Of particular interest, numerous investigations evaluate the potential of natural or synthetic compounds that stimulate apoptosis in breast cancer cells by disturbing intracellular Ca2+ homeostasis 3-9.
Ca2+ possesses an essential regulatory role in differentiation, secretion, contraction, transcription, phosphorylation, and apoptosis processes. A piece of large cell machinery composed of different proteins and organelles contributes to the regulation of intracellular Ca2+ concentration ([Ca2+]i) 10. Among them is the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), which allows active transport and a considerable accumulation of this cation at the endoplasmic reticulum (ER). Active transport maintains [Ca2+] in the ER. This concentration is approximately three orders of magnitude higher (millimolar range) than cytoplasm [Ca2+] (~100 nanomolar), and similar to the extracellular milieu11. Apoptosis is directly related to Ca2+ transfer from ER to mitochondria. Ca2+ depletion in ER can generate a phenomenon called “ER stress” in which the reduction of protein folding capacity, and consequent accumulation of these misfolded proteins, initiates different processes promoting cell death 12,13. The electrophoretic uniporter (MCU) induces Calcium accumulation in the mitochondria. This [Ca2+] increase generates an important change in electrochemical potential (Δψm) 14 which may enable membrane permeabilization with consequent release of cytochrome c, caspase-9 activation and cell death 15.
In this investigation, the effect of tetrahydroquinolines JS-56 and JS-92 (Fig. 1) was evaluated on Ca2+ homeostasis and apoptosis induction in breast cancer cells MCF-7. The tested compounds were prepared from inexpensive and commercially available isatin, 2-aminobenzonitrile (or 4-bromoaniline) and trans-isoeugenol, a major constituent of clove oil using a previously published twostep procedure 16,17. These tetrahydroquinolines have been shown to exert antitumor activity against various cancer cell lines, including MCF-7, SKBR3, PC3 and Hela cells (16, 17); the aim of the study is to ascertain the mechanism of apoptosis induction by the compounds. Both compounds induced an increase in [Ca2+]i caused in part through SERCA inhibition, combined with the dissipation of mitochondrial Δψm. Molecular docking analysis showed that these compounds have similar ligand-protein interactions with SERCA when compared to thapsigargin (Tg). Furthermore, these tetrahydroquinolines generated apoptosis via caspase activation and DNA fragmentation. Both compounds enhanced the effect of phorbol 12-myristate-13-acetate (PMA) on the NF-κB-dependent gene expression.

MATERIALS AND METHODS
Chemicals
The preparation of 8’-ciano-4’-(4-hydroxy-3-methoxyphenyl)-3’-methyl-3’,4’-dihydro-1’Hspiro[indoline-3,2’-quinolin]-2-one (JS-56) and 6’-bromo-4’-(4-hydroxy-3-methoxyphenyl)-3’- methyl-3’,4’-dihydro-1’H-spiro[indoline-3,2’- quinolin]-2-one (JS-92) was performed by following our previous reports which include the reaction of isatin and 2-aminobenzonitrile or 4-bromoaniline to give the respective ketimineisatin derivatives and the subsequent acidcatalyzed cycloaddition reaction with transisoeugenol 16,17. Both compounds were purified by silica gel column chromatography and obtained as stable solids. Their structures were confirmed by various spectroscopic techniques such as EI-MS, 1H NMR, 13C NMR, and IR, testing each sample as a pure chemical substance in the following biological experiments.
Cell culture
The human breast cancer cell line MCF-7 was grown in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with penicillin (100 U/mL), streptomycin (10,000 μg/mL), 1% GlutaMAX™ and 10% fetal bovine serum (FBS). These cells were kept in a humidified incubator at 37°C and in an atmosphere of 5% CO2.
Cytotoxicity assays
To evaluate the cytotoxicity in MCF-7 cells, the colorimetric reduction assay of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was performed 18. Ten thousand cells were seeded in 96-well plates for 48 h allowing confluence. Cells were exposed to JS-56 and JS-92 for 24 h, at concentrations ranging from 1 to 50 μM. In all cases, both tetrahydroquinolines were dissolved in dimethyl sulfoxide (DMSO). The final concentration of this solvent was equal to or less than 0.5%.
All appropriate controls were developed with 0.5% DMSO. After treatment, cells were incubated with 0.4 mg/mL of MTT for 2 h at 37°C. Then, the supernatant was removed, and the formazan crystals were dissolved by adding 50 μL of DMSO. Absorbance was measured in a multi-detection microplate reader (Sinergy HT, Bio-Tek) at 570 nm. The IC50 value was defined as the tetrahydroquinoline concentration causing a 50% reduction in absorbance compared to the control of untreated cells, using the Graphpad Prism 5.0 software.
Measurement of intracellular Ca2+ concentration
Measurements of [Ca2+]i in MCF-7 cells were made using the fluorescent indicator Fura-2, essentially as previously described 19. Briefly, a suspension of 1x106 cells/mL was prepared in a medium containing 138 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 5 mM glucose, 10 mM HEPES and 0.1% albumin (extracellular medium), loading cells with 200 μM sulfinpyrazone, 0.025% pluronic acid, 0.1% albumin, and 2 μM Fura 2 acetoxymethyl ester (Fura 2-AM) for 30 min at 25ºC, in darkness and constant agitation. After loading, cells were incubated for 15 minutes in the extracellular medium and washed again using the same solution. When indicated, these experiments were also performed using the same wash buffer but without CaCl2 (by addition of 2 mM EGTA). Fura-2 fluorescence on loaded cells suspension was monitored through a Perkin Elmer LS 55 spectrofluorimeter with a chopper device. The excitation wavelengths were set at 340 and 380 nm, while the emission was measured at 510 nm. All experiments were carried out at 30°C, with constant agitation in a stirred cuvette.
Intracellular Ca2+ was calculated according to Grynkiewicz et al.20, using the following equation: [Ca2+]I = Kd x (R-Rmin/Rmax-R) x Fmin(380)/Fmax(380) where Kd is the dissociation constant of Fura-2 (224 nM). R is the ratio of emission fluorescence intensities to excitation lengths of 340/380 nm. Rmin and Rmax are ratios at 0 Ca2+ (6 μM digitonin plus 4 mM EGTA) and saturating Ca2+ (2 mM CaCl2), respectively. Fmin(380)/Fmax(380) are fluorescent values of cells exposed to digitonin plus 4 mM EGTA and 2 mM CaCl2, representing minimal and maximal fluorescent values.
Purification of Ca2+-ATPase from the sarcoplasmic reticulum
The sarcoplasmic reticulum was obtained from the white skeletal muscle of the hind legs of rabbits, according to the method described by Eletr and Inesi 21. The vesicles derived from the sarcoplasmic reticulum are highly enriched in Ca2+-ATPase (approximately 90%). To avoid Ca2+ retention in the lumen of the vesicles, the ionophore A-23187 (1 μM) was added.
Evaluation of SERCA activity
The purified enzyme (2 μg/mL) was incubated for 45 min at 37°C and continuous agitation in a final volume of 250 μL of a buffer containing 100 mM KCl, 20 mM MOPSTris (pH 7.0), 1 mM EGTA, 10 mM MgCl2, 4 mM ATP, 1 μM A23817 and 1 mM CaCl2, obtaining 10 μM of Ca2+ in the medium and reaching an optimal activity of the enzyme. Inorganic phosphate production was quantified using the Fiske and Subbarow 22 colorimetric assay, modified by applying ferrous sulfate as a reducing agent 23.
Determination of the mitochondrial electrochemical potential (Δψm)
Mitochondrial membrane potential changes were measured using the fluorescent marker rhodamine 123, as previously described 24, with the modifications introduced by Pimentel et al.6. Rhodamine 123 is a cationic fluorescent dye that allows the evaluation of the electrochemical potential of this organelle when distributed according to the ∆ψ m. Rhodamine has a maximum excitation peak of 488 nm and a maximum emission peak of 530 nm. For these experiments 3 x 105 cells/mL were resuspended in a medium containing 138 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 5 mM glucose, 10 mM HEPES-KOH, pH 7.4 and then loaded with rhodamine 123 (20 μg/mL) for 45 min at 30ºC. All measurements were made on a HITACHI-L-7000 spectrofluorimeter at 30ºC with continuous stirring.
Determination of caspases 7 and 9 activity
We used Caspase-Glo 3/7 and Caspase-Glo 9 kits (Promega, Madison, WI) and developed the respective tests following the manufacturer’s instructions. Since MCF-7 cells do not possess caspase 3 25, the results shown correspond to the effect of the different compounds on caspase 7 activity. In a 96-well plate, 10,000 cells/well were seeded and incubated for 48 h at 37°C in a 5% CO2 atmosphere. Then, cells were exposed to different treatments for 24 h. Then, the cells were lysed, and the luminescence was measured with a multidetector microplate reader (Synergy HT, Bio-Tek). Staurosporine (Stau, 1 μM) was applied as an apoptosis activator (positive control), and 0.5% DMSO was used as a negative control.
Determination of DNA fragmentation
DNA fragmentation was analyzed through the “dead-end fluorimetric TUNEL (TdT-mediated dUTP Nick-End labeling) system” (Promega) kit. Cells were double-labeled with Fluorescein-12-dUTP (a specific marker of fragmented DNA) and propidium iodide (DNA marker). According to manufacturing instructions, changes in fluorescence intensity were detected by flow cytometry (Epics XL Beckman Coulter, FL). Cells incubated with staurosporine (1 μM) were used as a positive control.
Analysis of NF-κB-dependent gene expression
In this assay, HeLa tumor cells transfected with a luciferase reporter gene whose expression is under control of a modified IL-6 promoter, which only contains a critical binding site for NF-κB, were used. The luciferase activity was determined in a luminometer. Five thousand cells/well were seeded in 96-well plates and incubated for 48 h. Subsequently, the cells were exposed to all treatments and corresponding controls for 24 h. Then, the cells were lysed, the luciferase substrate was added, and the product was quantified by luminescence. Incubation with culture medium represents the negative control while phorbol 12-myristate 13-acetate (100 nM) was used as a positive control.
Molecular docking procedure
A 3D crystal structure from SERCA in complex with BHQ (2,5-ditert-butylbenzene-1,4-diol) and Tg was downloaded from Protein Data Bank under PDB code 2AGV. The protein corresponds to sarco(endo)plasmic reticulum Ca2+-ATPase from Oryctolagus cuniculus species, and it was crystallized with a resolution of 2.4 Å 26. The protein presented six co-crystallized chemical ligands: two molecules of BHQ, two molecules of Tg, and two sodium atoms. Four active sites (two for BHQ and the other two for Tg) were generated on the protein: two on α-chain and the other two on β-chain. The 3D structure was imported into the Swiss-PdbViewer v4.0.1 software 27. Hydrogen atoms were added to the protein structure, and minimization calculations were carried out on protein structure via AMBER force field, using the conjugate gradient method with an RMA gradient of 0.01 kcal/Åmol on Swiss-PdbViewer v4.0.1 software. The prepared protein was exported to an ArgusLab v.4.0.1 program package and saved as an Agl document. Four active sites were constructed on the SERCA protein derived from BHQ and Tg inhibitors. It is well documented that both compounds, BHQ and Tg, act as SERCA inhibitors. Specifically, BHQ is an L-type Ca2+ channel inhibitor, blocks Ca2+ transport by inducing superoxide anion production 28. On the contrary, Tg inhibits all SERCA isozymes at similar concentrations29. Both inhibitors are bound to the transmembrane domain of the enzyme close to the membrane/cytosolic interface.
Once the protein was prepared, the two tested tetrahydroquinolines JS56 and JS92 were built using ArgusLab v4.0 30, optimizing their respective geometries employing B3LYP/6-31G(d,p) 31. Then, molecular docking of the two compounds and crystallized inhibitors (BHQ and TG) over the four active SERCA sites was performed employing the ArgusLab (v4.0.1) package program under Windows 7.0 environment, applying AMBER force field. The docking was achieved through their respective grid map dimensions and a grid point spacing of 0.375 Å. A flexible ligand model was used in the docking and subsequent optimization scheme. Different orientations of the ligands were scanned and ranked from their energy scores. Reproducibility of the calculated affinity and the minimum energy pose was evaluated through 10 replicates for each quinoline and crystallized inhibitor (BHQ or TS) 32. The ChemScore scoring function was selected for the evaluation of ligand binding modes. Settings for genetic algorithm runs were kept at their default values; that is, the population size was 100, the selection pressure 1.1, and the number of operations was 100,000. Affinity energy is reported as the mean of the 10 replicates. The lowest energy poses (strongest-docking) for each ligand in each target protein are summarized in Table 1.
Table 1 Binding energies (kcal/mol), H-interactions, and hydrophobic interactions from molecular docking on the four studied active sites.
| Binding site | Molecule | Binding energy (kcal/mol) | H-interactions (Å) | Intermolecular interactions |
|---|---|---|---|---|
| TG1 | JS-56 | -13.1366 | Two hydrogen bonding (2.8596 and 2.1804 Å) between the phenolic hydrogen of the ligand and Ile-829 residue. |
a) H-π interaction with Phe-834 and Phe-256. b) Hydrophobic interactions with Met-838, Tyr-837, Phe-834, Ile-829, Leu-828, Pro-827, Leu-828, Ile-765, Gln-259, Leu-260 and Val-263. |
| JS-92 | -12.5189 | Hydrogen bonding (2.8988 Å) between the phenolic hydrogen of the ligand and Phe-834 residue |
a) H-π interaction with Phe-236 and Phe-834. b) Hydrophobic interactions with Met838, Ile829, Pro827, Leu828, and 765Ile. |
|
| Tg | -14.1626 | No hydrogen bonding |
a) No π-π interactions. b) Hydrophobic interactions with Met-838, Phe-834, Ile-829, Leu-828, Pro-827, Phe-776, Val-773, Val-772, Val-769, Asn-768, Ile-765, Ile-761, Pro-308, Ile-267, Val-263, Leu-260, Gln-259, Phe-256, Leu-253, Leu-249 residues. |
RESULTS
Effect of tetrahydroquinolines JS-56 and JS-92 on cytotoxicity in MCF-7 cells
First, the possible cytotoxicity generated by JS-56 and JS-92 on MCF-7 breast cancer cells was evaluated through an MTT assay after being treated for 24 h with different concentrations of these tetrahydroquinolines. It can be observed (Fig. 2) that both compounds induced cytotoxicity in MCF-7 cells in a dosedependent manner, with an IC50 of 9.74 μM and 5.03 μM for JS-56 and JS-92, respectively. At a concentration of 50 μM, JS-56 decreased cell viability by approximately 88%. At the same time, JS-92 exerted a more substantial effect than JS-56 on this cell line, being able to reduce almost 100% of MCF-7 cell viability at the same concentration.
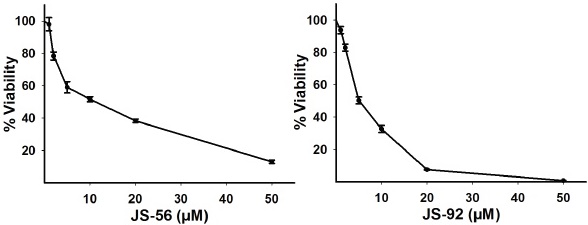
Fig. 2 Effect of tetrahydroquinolines JS-56 and JS-92 on MCF-7 cell viability. MCF-7 cells were treated with different concentrations of these compounds. Each point represents the mean ± SD of three independent experiments. IC50 values obtained for JS-56 and JS-92 were 9.74 μM and 5.03 μM, respectively.
Effect of tetrahydroquinolines JS-56 and JS-92 on intracellular Ca2+ mobilization
To study the effect of these tetrahydroquinolines on the [Ca2+]i, cells were loaded with the Ca2+ fluorescent indicator Fura-2. The addition of JS-56 induced a rapid increase in [Ca2+]i, reaching a peak followed by a continuous decrease (Fig. 3A), as a consequence of [Ca2+]i regulation carried out by the cellular homeostatic machinery until it reached a new steady-state Ca2+ level, which is greater than the original basal level. This response is characteristic of compounds capable of inducing the release of Ca2+ from the ER, with the consequent opening of plasma membrane Ca2+ channels (SOCE) activated by emptying this intracellular reservoir 19,33. The effect observed upon the addition of thapsigargin (Tg) (Fig. 3B), a potent SERCA inhibitor, was similar to that observed with JS-56. However, the maximum Ca2+ peak achieved with Tg was higher than JS-56. Additionally, when comparing Figures 3A and 3B, the addition of Tg after JS-56 (Fig. 3A) still induced a slight increase in [Ca2+]i. However, the tetrahydroquinoline did not exert any discernible effect after Tg addition (Fig. 3B).
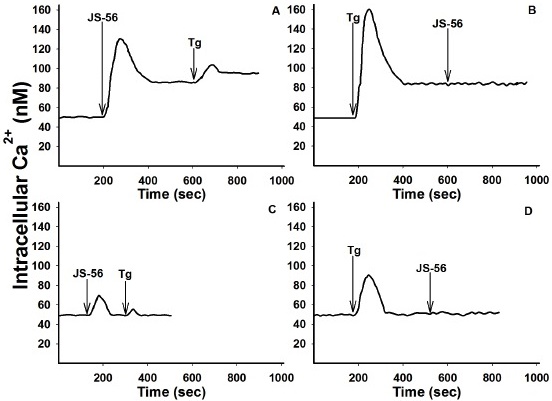
Fig. 3 Effect of tetrahydroquinoline JS-56 and thapsigargin (Tg) on [Ca2+]i in MCF-7 cells. The arrows indicate the moment in which JS-56 (20 μM) and Tg (2 μM) were added, in the presence (Fig. A and B) or absence (EGTA, Fig. C and D) of 2 mM CaCl2. The curves are representative of at least three independent experiments.
The same experiments were performed in the absence of extracellular Ca2+ to determine the origin of the cation increase. Under these experimental conditions, the maximum Ca2+ peaks obtained with JS-56 (Fig. 3C) or Tg (Fig. 3D) were considerably lower than
those reached in the presence of extracellular Ca2+ (Figs. 3A and 3B). Furthermore, the Ca2+ levels obtained, after the maximum peak and the subsequent decrease, were equal to the basal levels (Figs. 3C and 3D). Moreover, when Tg was added after JS-56, a minimal rise in [Ca2+]i was detected, indicating again that, different from Tg, the tetrahydroquinoline was not able to release all the Ca2+ from the ER (Fig. 3D). Likewise, the effect of the tetrahydroquinoline JS-92 was evaluated under the same experimental conditions, obtaining essentially the same overall results, as can be observed in Fig. 4.
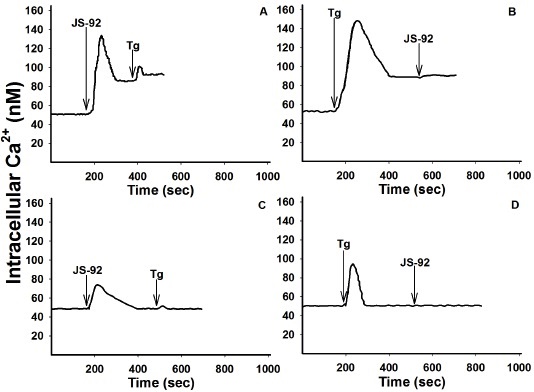
Fig. 4 Effect of tetrahydroquinoline JS-92 and thapsigargin (Tg) on [Ca2+]i in MCF-7 cells. The arrows indicate the addition of JS-92 (20 μM) and Tg (2 μM), in the presence (Fig. A and B) or absence (EGTA, Fig. C and D) of 2 mM CaCl2. The curves are representative of at least three independent experiments.
Effect of tetrahydroquinolines JS-56 and JS-92 on SERCA activity
To identify a possible mechanism of action of these compounds, which could explain their effect on the increase in [Ca2+]i, the ability to inhibit SERCA activity was examined, considering that the results obtained were very similar to those found by the use of Tg. Fig. 5 shows the effect of increasing concentrations of JS-56 and JS-92 on ATPase activity. Both tetrahydroquinolines inhibited SERCA activity in a dose-dependent manner. The maximum inhibitory effect of JS-56 and JS-92 on SERCA activity was 56 and 51%, respectively. Albeit none of the tetrahydroquinolines could completely inhibit the SERCA activity, these results demonstrate that part of the effect of these compounds on [Ca2+]i elevation was undoubtedly due to SERCA activity inhibition.

Effect of tetrahydroquinolines JS-56 and JS-92 on the mitochondrial electrochemical potential
Mitochondria use their electrochemical membrane potential (Δψm) to accumulate Ca2+ through the electrophoretic Ca2+ uniporter (MCU). The effects of the tetrahydroquinolines and Tg on the mitochondrial Δψ were studied by using the fluorescent marker rhodamine 123. This fluorescent indicator accumulates in the mitochondrial matrix according to the electrochemical gradient. Thus, an increase in fluorescence can be observed due to the mitochondrial H+ gradient dissipation, concomitantly with the release of rhodamine 123 from the mitochondria to the cytoplasm and then to the extracellular milieu. Results from Fig. 6 demonstrate that both JS-56 and JS-92 (20 μM) and Tg (2 μM) produced a rapid and pronounced elevation in fluorescence due to rhodamine 123 release from mitochondria. However, the increment in rhodamine 123 fluorescence caused by Tg was more significant than those produced by the tetrahydroquinolines (Figs. 6B and 6D). It can also be noticed that JS-92 (Fig. 6C) generated an elevation of rhodamine 123 fluorescence higher than JS-56 (Fig. 6A).
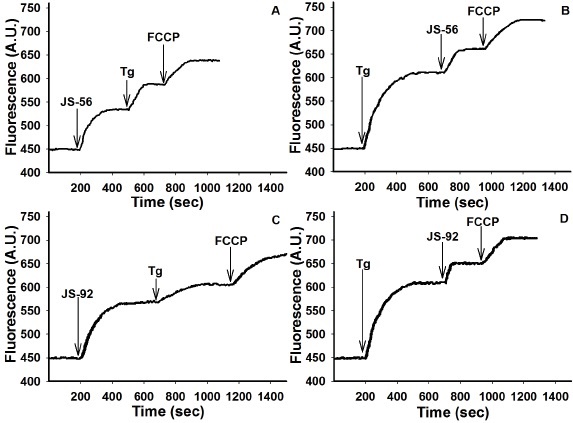
Fig. 6 Effect of tetrahydroquinolines JS-56 and JS-92 on the mitochondrial electrochemical potential in MCF-7 cells. The fluorescent marker rhodamine 123 was used for these experiments, expressing results in arbitrary units (AU) of fluorescence. The arrows indicate the moment of the addition of the different effectors: JS-56 or JS-92 (20 μM), thapsigargin (Tg, 2 μM), and FCCP (10 μM). The curves are representative of at least three independent experiments.
The effect of Tg after the addition of the tetrahydroquinolines was less pronounced (Figs. 6A and 6C). It is worthwhile to mention that this fluorescence elevation with both Tg and tetrahydroquinolines was expected since it was due to the partial dissipation of mitochondrial Δψ, generated by the entry of Ca2+ released from the ER6. On the other hand, after Tg application, the addition of JS-56 and JS-92 led to an additional boost in fluorescence (Figs. 6B and 6D), indicating a superior Δψ dissipation, which cannot be merely explained by the entrance of Ca2+ released from the ER. Thus, these results suggest that both tetrahydroquinolines can directly affect the mitochondria by Ca2+-independent mechanisms. As expected, the positive control, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), generated a complete collapse of the electrochemical gradient.
Effect of tetrahydroquinolines JS-56 and JS-92 on the activity of caspases 7 and 9
We then evaluated the possible effect of these tetrahydroquinolines on the induction of apoptosis in MCF-7 cells. Accordingly, the activation of caspase 9, a known caspase that initiates apoptosis, was assessed. The 1/2 IC50, IC50, and 2 IC50 obtained in the MTT assay were used for both compounds. As shown in Fig. 7 (upper panel), both JS-56 and JS-92 significantly enhanced the activity of caspase-9 compared to the control in a dose-dependent manner. Sphingosine (Sph), which promotes an increase in [Ca2+]i in MCF-7 cells 33, and 2,5-di-t-butyl-1,4-benzohydroquinone (BHQ), a known SERCA reversible-inhibitor 34, also elevated the activity of this initiating caspase. The positive control Stau, which activates apoptosis in MCF-7 cells 35, effectively activated caspase 9 at a level similar to the IC50 of the tetrahydroquinolines.
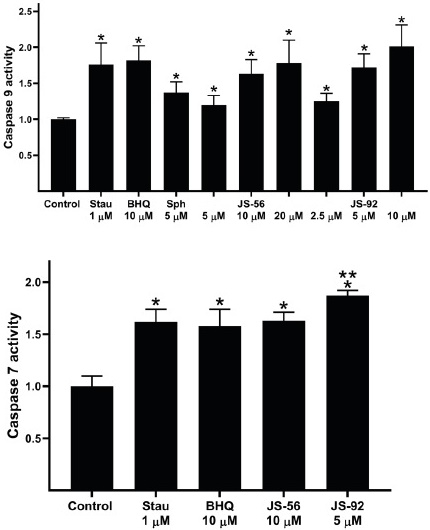
Fig. 7 Effect of tetrahydroquinolines JS-56 and JS-92 on the activity of caspases 9 (initiator) and 7 (effector). Stau (staurosporine), BHQ (2,5-di-t-butyl-1,4-benzohydroquinone) and Sph (sphingosine). The bars represent the mean ± SD derived from three independent experiments. * and ** p < 0.05 compared to the control of untreated cells or treated with JS-56, respectively.
Likewise, treatment of MCF-7 cells with both tetrahydroquinolines raised the activity of caspase 7 (Fig. 7, lower panel). Similar results were detected with the positive control Stau and the SERCA inhibitor BHQ. The activation-induced by JS-92 was slightly higher compared to the rest of the compounds. This effector caspase will be responsible for cleaving specific cell targets to promote apoptosis in these cells.
Effect of tetrahydroquinolines JS-56 and JS-92 on DNA fragmentation
An apoptotic cell is typically characterized by: changes in cell morphology, phosphatidylserine externalization, cytoplasm contraction, shrinkage of the nucleus, and DNA fragmentation 15. As a complement to the results obtained in caspases 7 and 9 activity assays, DNA fragmentation was analyzed through a TUNEL assay using Stau as a positive control. In Fig. 8, cells were divided into four populations displaying the percentage of cells in necrosis (propidium iodide, IP+), apoptosis (fluorescein isothiocyanate, FITC+), and alive (negative for both markers); apoptotic cells can either be IP- or IP+. Apoptotic cells exhibit DNA fragmentation, which allows FITC incorporation. However, a subset of these apoptotic cells is also IP+, denoting that these also have a loss of plasma membrane integrity, corresponding to a later stage of apoptosis in which cells can no longer control the passage of elements across the membrane, with eventual cell lysis in the final stage. The quantitative data is represented in Table 2.
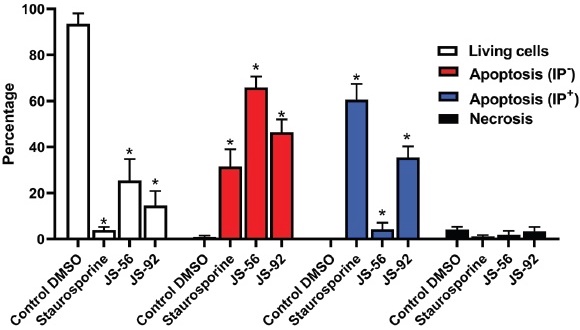
Fig. 8 Effect of tetrahydroquinolines JS-56 and JS-92 on DNA fragmentation in MCF-7 cells. The MCF-7 cells were incubated with Fluorescein-12-dUTP (fragmented DNA marker) and Propidium Iodide (IP, core marker). Cells were treated with staurosporine (1 μM), JS-56 (10 μM), and JS-92 (5 μM). The bars represent the mean ± SD derived from three independent experiments. * p < 0.0001 compared to the control of cells treated with DMSO.
Table 2 Percentage of living, apoptotic, and necrotic cells after treatment with JS-56 and JS-92.
| Treatment group | Percentage | |||
|---|---|---|---|---|
| Living cells | Apoptosis (IP-) | Apoptosis (IP+) | Necrosis | |
| Control DMSO | 93.5 ± 4.6 | 0.9 ± 0.6 | 0.1 ± 0.1 | 4.1 ± 1.2 |
| Staurosporine | 3.9 ± 1.3 | 31.5 ± 7.5 | 60.6 ± 6.8 | 1.2 ± 0.5 |
| JS-56 | 25.5 ± 9.3 | 65.9 ± 4.7 | 4.3 ± 2.8 | 1.9 ± 1.6 |
| JS-92 | 14.6 ± 6.3 | 46.5 ± 5.5 | 35.5 ± 4.8 | 3.4 ± 1.8 |
JS-56 treatment (10 μM) for 24 h induced apoptosis, with 65.9% of FITC+ IPcells in this stage and only 4.3% of FITC+ IP+ cells. Treatment with Stau (1 μM) and JS-92 (5 μM) produced a significant percentage of FITC+ IP- (31.5 and 46.5% for Stau and JS-92, respectively) and FITC+ IP+ cells (60.6 and 35.5% for Stau and JS-92, respectively). Hence, JS-92 has a more potent effect than JS-56, which measurements of caspase activity have also evidenced. These results are statistically significant compared to the negative control (DMSO-treated MCF-7 cells).
Effect of tetrahydroquinolines JS-56 and JS-92 on NF-κB-dependent gene expression
The nuclear transcription factor NF-κB regulates numerous genes involved in multiple cellular processes such as apoptosis, cell proliferation, and differentiation. To evaluate NF-κB-dependent gene expression, HeLa tumor cells transfected with a luciferase reporter gene under the control of IL-6 promoter, which contains critical binding sites for NF-κB, were used.
Tetrahydroquinolines JS-56 and JS-92, when added alone, were not able to increase the NF-κB-dependent gene expression, with values similar to those observed in the negative control (Fig. 9). The luminescence measured in the negative control corresponds to the basal NF-κB-dependent gene expression on MCF-7 cells. The NF-κB-dependent gene expression is elevated when cells were treated with 100 nM of PMA; it has been reported that PKC can activate IκBs kinase complexes (IKKs), which eventually leads to NF-κB activation 36. Interestingly, the results demonstrate that the NF-κB-dependent gene expression increased significantly when cells were treated with PMA plus JS-56 or JS-92, indicating an apparent effect of both compounds on the PKC-stimulated NF-κBdependent gene expression.
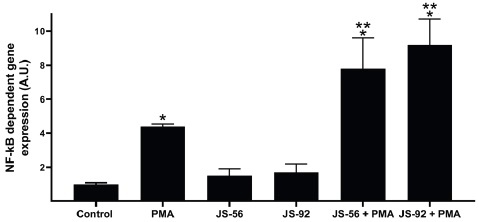
Fig. 9 Effect of tetrahydroquinolines JS-56 and JS-92 on NF-κB-dependent gene expression. HeLa tumor cells were transfected with a luciferase reporter gene under the control of the IL-6 promoter, which contains critical binding sites for NF-κB. Control, untreated MCF-7 cells; PMA, phorbol 12-myristate 13-acetate. * and ** p<0.05 compared to the control of untreated cells or treated with PMA, respectively.
Molecular docking
The experimental results on SERCA activity obtained for the tetrahydroquinolines JS-56 and JS-92 were confirmed using molecular docking for SERCA protein (PDB code 2AGV). Interestingly, both Tg and BHQ SERCA inhibitors showed practically the same disposition as crystallized inhibitors at the respective protein binding sites, reflecting the computational package’s potential for this molecular docking. In general, the tetrahydroquinolines showed a higher preference for Tg binding sites than for the BHQ active site. This effect generates a ligand-protein complex with binding energies between -12 and -13 kcal/mol at the active sites of Tg; this is in contrast to weak docking energies found at the BHQ active site for both tetrahydroquinolines. Particularly, experimental results have demonstrated that Tg exerted a more potent inhibitory activity (IC50 of 0.2 nM) than BHQ (IC50 of 20 nM) 26. The preference of these large tetrahydroquinolines for Tg binding sites may be associated with the larger box size than BHQ active sites, which may facilitate the stabilization of the inhibitor-protein complex for these bulky and inflexible molecules. It is essential to mention that our analysis was focused on data obtained for the Tg active site from chain-α, which exhibited the lower binding energy complex.
DISCUSSION
Tetrahydroquinolines derivatives are an essential group of natural or synthetic compounds showing a wide range of biological activities. Numerous compounds capable of inducing a Ca2+ signal followed by a cellular response have been reported. Ca2+ is one of the essential ionic constituents in the body. It has a central regulatory role in differentiation, secretion, contraction, transcription, phosphorylation, and apoptosis processes 10. Ca2+ is the component required by milk to calcify newborn bones and teeth and modulates the proliferation, differentiation, and apoptosis of mammary gland epithelial cells 37. The modulation of Ca2+ homeostasis and its signaling could represent a viable therapeutic approach in the treatment and/or prevention of breast cancer. Therefore, regulators of Ca2+ homeostasis or its signaling in mammary gland tumors are possible targets for drugs. In this investigation, we used the tetrahydroquinolines JS-56 and JS-92, modified derivatives from clove and cinnamon plants, to study the effect of these drugs on Ca2+ homeostasis and its possible relationship with apoptosis in breast cancer cells MCF-7.
In the interest of evaluating the cytotoxicity of tetrahydroquinolines JS-56 and JS-92 on MCF-7 breast cancer cells, the MTT assay was developed, allowing the determination of mitochondrial viability on treated cells. Both compounds are cytotoxic to MCF-7 cells in a dose-dependent manner, with an IC50 of 9.74 μM and 5.031 μM for JS-56 and JS-92, respectively. This difference may be related to the bromine atom that JS-92, instead of the cyanide atom present in JS-56. IC50 values for these compounds in MCF-7 cells are significantly lower than those reported previously in human dermis fibroblast, 92.87 µM and 42.48 µM for JS-56 and JS-92, respectively 17.
Through fluorimetric techniques, we were able to perform measurements of intracellular Ca2+ in MCF-7 cells, aiming to find possible mechanisms of action for these compounds. The results revealed that JS-56 and JS-92 promote an increase in [Ca2+]i. Likewise, the Tg effect on [Ca2+]i is diminished when added after the impact generated by the tetrahydroquinolines, compared to when it is initially added, allowing us to conclude that these compounds promote the release of Ca2+ from a compartment sensitive to Tg, namely the ER. Similar experiments were done but using a solution without CaCl2. Under these experimental conditions, the maximum Ca2+ peaks detected with JS-56, JS-92 or Tg are considerably reduced compared to those achieved in the presence of extracellular Ca2+, and instead, returning to the basal level. These results show an influx of Ca2+ from the outside. This effect may be caused by a store-operated Ca2+ Entry (SOCE) or a capacitive Ca2+ entry induced by emptying Ca2+ from the ER 19,33.
The lack of an additive effect of Tg with tetrahydroquinolines on the [Ca2+]i suggests a possible similar mechanism of action. The possible inhibitory potential of these compounds on SERCA activity was examined accordingly. Both tetrahydroquinolines inhibit the enzyme activity with similar maximum inhibitory effects of 56 and 51% for JS-56 and JS-92, respectively. This effect also indicates that different from Tg, these compounds could not completely inhibit the ATPase activity. However, part of the overall effect on the onset of apoptosis is the partial inhibition of SERCA activity. As it has been shown that Tg can induce ER stress via SERCA inhibition, JS-56 and JS-92 are likely potential candidates for generating ER stress through this identical mechanism. In this context, it is worth mentioning that SERCA targeting could represent an effective therapeutic strategy in treating pathogens and cancer cells 38.
The effect of both compounds on the mitochondrial membrane Δψm was determined since the accumulation of mitochondrial Ca2+ is a necessary event in the initiation of apoptosis via the intrinsic pathway. The primary source of the cation derives from its transfer from the ER 39. The results revealed that both tetrahydroquinolines generate an increase in rhodamine 123 fluorescence. Although the increment in rhodamine fluorescence caused by Tg was higher than the compounds, its effect after the addition of the tetrahydroquinolines was less pronounced, suggesting that they share part of the mechanism of action on mitochondria. The fluorescence increase after treating the cells with both Tg and tetrahydroquinolines was expected due to the partial dissipation of mitochondrial Δψm. This change is generated by the entry of Ca2+ released from the ER 6. When both tetrahydroquinolines were added after Tg, an additional increase in fluorescence was triggered, indicating a superior dissipation of the Δψ. These unexpected results suggested that JS-56 and JS-92 induced a cell response independent of [Ca2+]i. The effects are also independent of the Ca2+ electrophoretic uniporter. These effects have been reported previously with concentrations higher than 1 µM 40. Accordingly, both tetrahydroquinolines, similar to the SERCA inhibitor, could interact with the mitochondria, generating a widespread effect.
Given that the Δψm loss of the mitochondrial membrane is a hallmark event of apoptosis, the presumed activation of characteristic processes associated with programmed cell death after applying the tetrahydroquinolines was investigated. In this study, it was demonstrated that tetrahydroquinolines JS-56 and JS-92 are capable of activating caspases 7 and 9 and promoting DNA fragmentation. Activation of caspases and the percentage of cells with DNA fragmentation was higher for JS-92, which coincides with its superior cytotoxic effect. DNA fragmentation in conjunction with caspase activation and the results demonstrating that these tetrahydroquinolines cause the dissipation of mitochondria Δψm to support the conclusion that these compounds activate apoptosis in MCF-7 cells. Additionally, after the inhibition of SERCA and the consequent reduction of the [Ca2+] at the ER, misfolded proteins could accumulate. The unfolded protein response could be triggered, leading the cells to death (12, 13). By suppressing this enzyme’s activity and promoting the cation’s entry into the mitochondria, the release of pro-apoptotic factors from this organelle would be facilitated 15. Thus, part of the potential anticancer effect of these compounds would be mediated through SERCA inhibition. Based on the accumulated evidence, we suggest that disturbances of the Ca2+ signal induced by these tetrahydroquinolines would trigger apoptosis by promoting ER stress and the release of proapoptotic factors from the mitochondria after the entry of Ca2+ into this organelle.
According to our results, JS-56 and JS-92 failed to elevate the NF-κB-dependent gene expression. However, NF-κB-dependent gene expression was augmented when cells were treated with PMA, an activator of PKC which upregulates NF-κB 35. Interestingly, the NF-κB-dependent gene expression is further elevated when cells were exposed to the combination of PMA and tetrahydroquinolines, possibly because these compounds enhance the affinity of PKC for PMA or modulate the PMA activity making it more potent and/or stable. Since NF-κB is a regulator of the expression of multiple genes, additional experiments are needed to determine its putative participation in pro-apoptotic events. We do not rule out non-specific binding of other transcription factors to this NF-κB binding site on the IL-6 promoter. Future research will confirm the participation of this transcription factor by determination of cytoplasmic/nuclear levels of p65 as well as confirmation of these results in breast cancer cells.
Based on the results that both tetrahydroquinolines may interact with the Tg binding site of SERCA, we used a docking model to test the possibility of interaction. It should be noted that both tetrahydroquinolines exhibited docking energies higher than Tg, indicating that the Tg-Ca2+-ATPase complex is more stable than the quinoline-Ca2+-ATPase complex (Table 1). These theoretical findings are in good agreement with experimental data, where Tg exhibited the higher experimental inhibitory activity (IC50 of 0.2 nM) compared to the tested tetrahydroquinolines (IC50 of 60 and 100 μM for JS-56 and JS-92, respectively). The high stabilization of the Tg-Ca2+-ATPase complex can be associated with diverse intermolecular interactions with residues located in the Tg1 active site, including (i) a large number of hydrophobic interactions with at least 20 residues in chain A and 13 residues in chain B; (ii) from one to two strong H-interactions with Ile-829 and Phe-834 for JS-56 and JS-92, respectively; and (iii) H-π interactions between phenolic, quinolinic, or indolic rings with specific phenyl rings in Phe-834, Phe-256 or Tyr-837 (Table 1 and Fig. 10). However, it is essential to mention that these diverse interactions found for these tetrahydroquinolines are located in turn to a specific region of the active site. At the same time, Tg, bearing a highly flexible octanoic acid residue at C2, makes hydrophobic contacts with non-polar patches on the outer surface of SERCA’s trans-membrane domain and possibly with surrounding lipids, facilitating the stabilization of the ligand-protein complex. The molecular docking analysis shows that hydrophobic ligands containing long and flexible side chains are needed to design effective inhibitors of the Ca2+-ATPase enzyme due to the high hydrophobic and extended nature of the Ca2+-ATPase active site. Future studies will continue to evaluate these types of compounds and their effect on Ca2+ and apoptosis, developing new analogs with different types of chemical modifications on a more diverse panel of breast cancer cells and other types of malignancies.
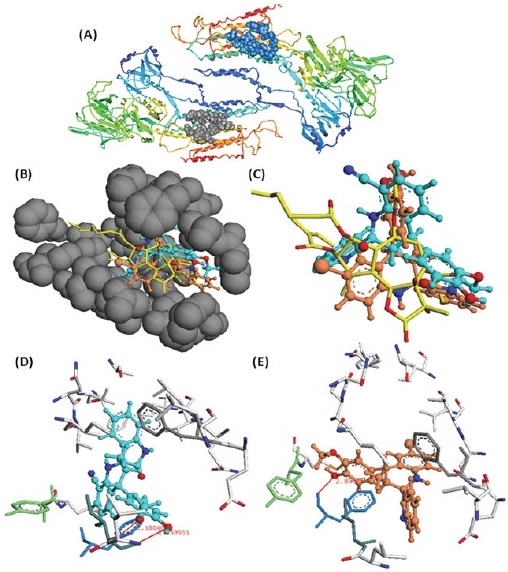
Fig. 10 Molecular docking of the tetrahydroquinolines JS-56 and JS-92 on the TG1 active site. (A) TG1 (gray ribbons) and TG2 (blue ribbons) active sites into SERCA protein; (B) Location of JS-56 (cyan color) and JS-92 (orange color) into the TG1 active site pocket of SERCA protein; (C) Superimposition between JS-56 (cyan) and JS-92 (orange) with TG1 substrate (yellow color); (D) Disposition and interactions of JS-56 (cyan) (D) and JS-92 (orange) (E) into TG1 active site. Tentative H-π interactions between phenolic, indolic, or quinolinic rings of JS-56 and JS-92 with Phe-236 (gray color), Phe-834 (blue color), and Phe-256 (green color) residues.
Both tetrahydroquinolines derivatives, JS-56 and JS-92, induced cell death in MCF-7 cells through a combination of events. These events include the elevation of the [Ca2+]i, inhibiting SERCA via direct interaction, partially damaging the mitochondria, which induced activation of apoptotic pathways and consequently MCF-7 cell death. Therefore, these results show the potential that these types of compounds would have in anti-neoplastic treatment in humans. It is hoped that more powerful analogs can be developed in the future.

