










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Oncología
versión impresa ISSN 0798-0582
Rev. venez. oncol. v.21 n.1 Caracas mar. 2009
Histiocitoma fibroso maligno pleomórfico. Presentación de un caso y revisión de la literatura
Carlos E Quintero R, Gabriela M González P, José L Valderrama
Unidad de Cirugía General Iahula, Cirugía General Hospital Patrocinio Peñuela Ruiz IVSS.
Correspondencia: Dra. Gabriela Margarita González Paredes. Unidad de Cirugía General. Instituto Autónomo Hospital Universitario de Los Andes. Av. 16 de Septiembre Sector Campo de Oro, Mérida, Venezuela Código Postal: 5101. Telf.: 0058-414-7473280, 0058- 416-3703766 E-mail: gabipar10@hotmail.com
RESUMEN
Se presenta un caso clínico de histiocitoma fibroso maligno pleomórfico diagnosticado mediante valoración clínica, tomografía axial computarizada con contraste, biopsia por tru-cut preoperatoria y estudio inmunohistoquímico e histológico. A quien en vista del tamaño, grado de diferenciación tumoral y ausencia de metástasis se decide realizar exéresis quirúrgica de la tumoración de 4 300 g, 50 cm x 30 cm y tratamiento radioterápico adyuvante. Evidenciándose satisfactoria evolución a sus seis meses de seguimiento, caracterizado por cicatrización estética y funcional, aumento de peso y ausencia de recidiva tumoral. En vista que la presentación de esta patología es poco frecuente en el mundo entero, se decide hacer una revisión de la literatura y discusión de los hallazgos anatomopatológicos y clínicos concernientes al caso.
PALABRAS CLAVE: Sarcoma, histiocitoma fibroso maligno, pleomórfico.
SUMMARY
Clinical case of patient with fibroses pleomorphic hystiocitome diagnosed by clinic valuation, on-line axial tomography with contrast, immunohystochemistry and pathology study. Who in view of the size, grade of differentiation tumor and metastasis absence decides to carry out a surgical exeresis of tumor 4 300 g, with a length 50 cm x 30 cm and radiotherapy coadjutant therapy. Being evidenced, satisfactory evolution six months of pursuit, characterized by aesthetic and functional scaring, increase of weight and absence of relapse tumor. In view of the fact that the presentation of this pathology is not very frequent in the whole world, we decide to make a revision of the literature and discussion of the discoveries pathology and clinical concerning to the case.
KEY WORDS: Sarcoma, malignant fibrose hystiocitome, pleomorphic.
Recibido: 30/01/2008 Revisado: 22/05/2008 Aceptado para publicación: 27/08/2008
INTRODUCCIÓN
Los sarcomas de partes blandas constituyen el 1 % de todas las neoplasias en el hombre (1), La derivación del tejido neoplásico está relacionada con el tejido mesenquimático y pueden tener cualquier localización, aunque aproximadamente el 50 % aparecen en extremidades. Representan una de las condiciones de mayor agresividad entre las neoplasias malignas (2).
OBrien y Stout (3) en 1964, fueron los primeros en describir el histiocitoma maligno. Actualmente esta neoplasia ha sido considerada el sarcoma de partes blandas más frecuente en adultos (4). No obstante, estudios recientes han demostrado que se pueden clasificar en función de su diferenciación, aportando por tanto distintas características clínicas y pronósticos (5). Es más frecuente sobre los 50 años, pero no hay una edad específica, puede aparecer a lo largo de toda la vida (2).
Desde el punto de vista macroscópico el histiocitoma fibroso maligno aparece como una tumoración grande con áreas múltiples de necrosis en la superficie de corte. Microscópicamente el tumor muestra una proliferación desordenada de células fusocelulares con ocasional patrón esteliforme o arremolinado, presencia de células multinucleadas con gran atipia nuclear, formas bizarras, frecuentes figuras de mitosis atípicas, y un estroma que muestra gran cantidad de colágeno así como un número variable de células inflamatorias mononucleadas e histiocitos espumosos (6).
Su diagnóstico es clínico y paraclínico. Cruz (6), afirma que en un 74 % de los casos, es realizado en base a su morfología macro y microscópica, pero aún así recomienda el uso de la inmunohistoquímica, la cual es considerada por Miettinen (7), fundamental por causa de la heterogenicidad de estas lesiones.
Valdés (8), manifiesta que el tratamiento de elección es la cirugía con márgenes de resección amplios, siendo la radioterapia y quimioterapia tratamientos complementarios de la cirugía. Sin embargo, otros autores (9), consideran que el tratamiento multidisciplinario es esencial por dos razones: 1. Porque el pronóstico de estas neoplasias está dado por el grado histológico y el tamaño del tumor, y 2. Porque hasta un 22 % de los casos presentan ya desde un inicio enfermedad clínica metastásica.
Lewis (17) afirma que la radioterapia adyuvante reduce significativamente el índice de recidivas locales. La braquiterapia reduce la incidencia de recidivas locales de un 31 % a 18 %, a dosis de 45 Gy.
CASO CLÍNICO
Paciente masculino de 51 años de edad, con tumoración en región escapular izquierda de 14 meses de evolución, ésta fue aumentando progresivamente de volumen, no refiere antecedentes personales ni familiares de importancia.
Al examen físico, se evidencia tumoración de 50 cm x 30 cm, que ocupa desde la región escapular izquierda hasta la región lumbar ipsilateral, bien delimitada, regular, blanda, no renitente, dolorosa a la palpación profunda, inmóvil, hipervascularizada (Figura 1 y 2). Sin palparse adenopatías.
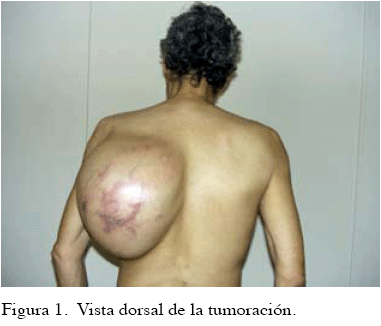
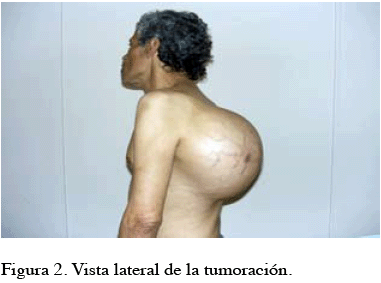
EXÁMENES COMPLEMENTARIOS
Analítica: Leucocitos: 15 730 x cm3 neutrófilos: 76,8 %; linfocitos: 15,5 %; monocitos: 6,7 %; Hb: 12,7 g/dL; plaquetas: 611 000 x cm3; glicemia: 109 mg/dL; creatinina: 0,6 mg/dL; TGO: 15 U/L; TGP: 16 U/L; Proteínas totales 7,2 g/dL; albúmina; 3,2 g/dL; globulinas: 4g/dL; TP: 13,8/12 seg; TPT:32,1/29,7 seg; VDRL y HIV no reactivos.
Tomografía torácica axial computarizada con contraste: Imagen dishomogénea redondeada de bordes definidos, la cual realza parcialmente posterior a la administración de contraste endovenoso (Figura 3.A y B). Biopsia por tru-cut preoperatoria: Neoplasia maligna mesenquimática, cuyas extensas áreas de necrosis tumoral, dificultan la realización de diagnóstico de certeza. Inmunohistoquímica preoperatoria: CD68 positivo, vicentina, citoqueratina y desmina negativos, concluyendo: histiocitoma fibroso maligno pleomórfico.

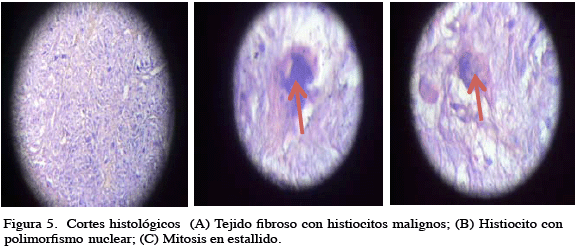
El paciente es llevado a la mesa operatoria realizándose exéresis total de la lesión, que pesó 4 300 g, sin evidencia macroscópica de compromiso en los planos profundos o escapular (Figura 4. A, B), con una evolución posoperatoria satisfactoria egresando al segundo día del posoperatorio. La biopsia escisional reportó: fibrohistiocitoma maligno tipo pleomórfico, pobremente diferenciado, con áreas de patrón fascicular, células xantomatosas, células inflamatorias, con pseudocápsula indemne y márgenes libres, motivo por el cual se indica radioterapia adyuvante. (Figura 5. A, B, C).
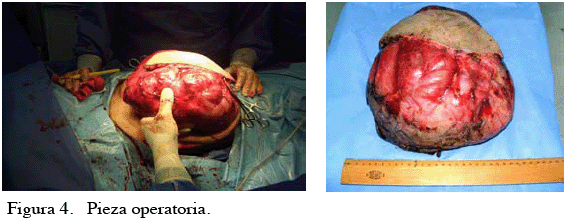
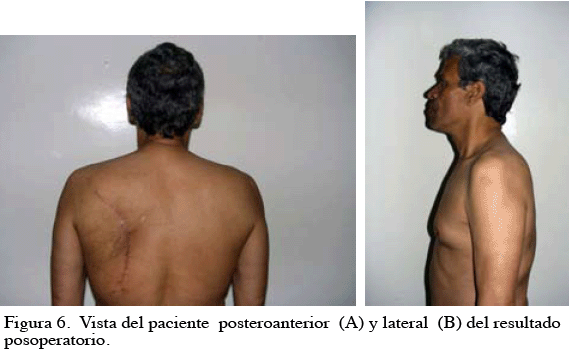
En las consultas de control se evidenció una cicatrización adecuada, con satisfactorio resultado estético y funcional (Figura 6. A y B).
DISCUSIÓN
Alfeirán (1), afirma que el 5 % de los sarcomas de partes blandas se localizan en pared torácica. La edad de aparición de esta lesión, fue similar a la reportada como la más frecuente en las series de Belal (10) y Kearney (11). La presentación clínica de esta tumoración indolora con crecimiento rápido y afectación vascular superficial, está descrita (12) como la más común. Sin embargo, existen otras formas menos frecuentes: masa dolorosa de aparición repentina y síntomas neurológicos o vasculares, relacionados con la localización y origen del tumor (8).
Los protocolos de estudios existentes para diagnóstico de sarcomas de partes blandas, estipulan la realización de estudios imaginológicos como tomografía axial computarizada con contraste, que permite establecer la localización, extensión, afectación de estructuras y tejidos adyacentes, así como las lesiones corticales, reacciones periósticas y matrices mineralizadas. Teniendo presente que no sirven para diferenciar los tumores benignos de los malignos, únicamente pueden determinar el patrón de agresividad (8). En el caso presentado se realizó TAC contrastada, la cual permitió establecer muchos de los criterios anteriormente mencionados.
La evaluación clínica se apoya en estudios de laboratorio para valorar la extensión del tumor, todo esto, a su vez, apoyado con el diagnóstico histopatológico, los tipos de biopsia más utilizados en sarcomas son: con aspiración por aguja fina, con tru-cut, incisional y escisional (15). La biopsia por tru-cut representa un método sencillo, con el cual se puede obtener material suficiente para llegar al diagnóstico histopatológico, con una sensibilidad de 94 % a 98 % (16).
Los estudios morfológicos macro y microscópico fueron fundamental para el diagnóstico, pero aún así se realizó inmunohistoquímica, con CD68 positivo, como lo sugiere Cruz (6) y Miettinen (7).
En el MD Anderson Hospital (12), en pacientes con sarcomas de tejidos blandos, etapas clínicas II y III, se aplicó exitosamente la estrategia de cirugía más radioterapia posoperatoria, con una supervivencia absoluta de 61,5 %.
El protocolo del Hospital de Basurto (8), para tratamiento de sarcomas de tejidos blandos, propone que los márgenes deben ser de 5 cm, aunque se acepta hasta de 1 cm si la fascia que recubre el tumor es sana. Debe evitarse incidir la pseudocápsula tumoral. La radioterapia está indicada en histiocitomas de medio y alto grado, bajo grado con márgenes inadecuados de resección, o aquellos casos en los que la recidiva requiera de amputación. La histopatología reportó: histiocitoma fibroso maligno pleomórfico, pobremente diferenciado, con pseudocápsula indemne y márgenes libres, lo que cataloga esta tumoración como de alto grado, motivo por el cual se indica radioterapia adyuvante.
La quimioterapia complementaria no debe ser una rutina, deben discutirse con el paciente las ventajas (aumento del tiempo libre de recaída local) e inconvenientes del tratamiento, dado el escaso beneficio esperable sobre la sobrevida (13).
Los factores pronósticos más importantes para los sarcomas de partes blandas son: tamaño tumoral, grado de diferenciación e invasión ganglionar o metastásica (14).
REFERENCIAS
1. Alfeirán A, Herrera A, Escobar G. Sarcomas de la pared torácica. Resección y reconstrucción. Rev Inst Nal Cancerol. 1997;43(4):189-193. [ Links ]
2. Mahiques A. Sarcomas de partes blandas. 2004. Disponible en: URL: http://www.arturomahiques.com/sarcomas_de_partes_blandas.htm [ Links ]
3. OBrien JE, Stout AP. Malignant fibrous xanthomas. Cancer. 1964;17:1037-1093. [ Links ]
4. Bertoni F, Capanna R, Biagini R. Malignant fibrous histiocytoma of soft tissue: An analysis of 78 cases located and deeply seated in the extremities. Cancer. 1985;56:356-367. [ Links ]
5. Fletcher CD, Gustafson P, Rydholm A, Willen H, Akerman M. Clinic pathologic re-evaluation of 100 malignant fibrous histiocytomas: Prognostic relevance of sub classification. J Clin Oncol. 2001;19(12):3045-3050. [ Links ]
6. Cruz J, Martínez I, Caballero I, Pérez L. Análisis del término histiocitoma fibroso maligno pleomórfico. Un estudio de los sarcomas pleomórficos de partes blandas. Rev Cubana Oncol. 1999;15(1):8-12. [ Links ]
7. Miettinene M. Immunohistochemistry of soft-tissue tumors. Possibilities and limitations in surgical pathology. Pathol Annu. 1986;1:1-36. [ Links ]
8. Valdés C, Oleada L, López I, Puertas J, Egilior J, Ortiz J, et al. Protocolo para diagnóstico y tratamiento de sarcomas de tejidos blandos del Hospital de Basurto. Cirugía Plástica Ibero-latinoamericana. 2004;30(4):285-292. [ Links ]
9. Escovar G, Herrera A. Diagnóstico y tratamiento de los sarcomas de partes blandas. Rev Inst Nal Cancerol. 1997;43(4):172-178. [ Links ]
10. Belal A, Kandil A, Allam A, Khafaga Y, El-Husseiny G, El-Enbaby A, et al. Malignant fibrous histiocytoma. A retrospective study of 109 cases. Am J Clin Oncol. 2002;25(1):16-22. [ Links ]
11. Kearney MM, Soule EH, Ivins JC. Malignant fibrous histiocytoma. A retrospective study of 167 cases. Cancer. 1980;45:167-178. [ Links ]
12. Lindberg R. Treatment of localized soft tissue sarcomas in adults at MD Anderson Hospital and Tumor Institute. Cancer. 1985;3:59-65. [ Links ]
13. U.K. Medical Research Council. Review: Adjuvant chemotherapy improves recurrent- free intervals and recurrent- free survival in soft tissue sarcomas. Oxford. 1998. [ Links ]
14. AJCC Cancer Staging Manual. American Joint Comittee on Cancer. 5a edición. Filadelfia: Lippincott-Raven; 1997. [ Links ]
15. Kissin M, Fisher C, Webb AJ, Westbury G. Value of the needle aspiration cytology in the diagnosis of the soft tumors: A preliminary study of the excised specimen. Br J Surg. 1987;74:479-480. [ Links ]
16. Kissin MW, Fisher C, Carter RL, Horton LW, Westbury G. Value of trucut biopsy in the diagnosis of the soft tissue tumors. Br J Surg. 1986;73:742-744. [ Links ]
17. Lewis J. The asociation of local recurrence with the subsequent survival in extremity soft tissue sarcoma. Abstract book, 49 Cancer Symposium. Society of Surgical Oncology. March 21 - March 24, 1996. [ Links ]
