










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalus
versión impresa ISSN 1316-7138
Salus vol.18 no.3 Valencia dic. 2014
Displasia fibrosa craneofacial avanzada por remodelación ósea.
Mathias Martínez Coronel, Héctor Rojas Mercado
Departamento de Estomatoquirúrgica. Cirugía Bucal. Universidad de Carabobo, Campus Bárbula. Venezuela.
Autor de correspondencia: Mathias Martínez Coronel E-mail:
RESUMEN
La displasia fibrosa es un trastorno del desarrollo esquelético del mesénquima de formación ósea que se manifiesta como un defecto en la maduración y diferenciación de los osteoblastos. La displasia fibrosa representa alrededor de 2.5% de todos los tumores óseos y más del 7% de los tumores benignos. El objetivo de este artículo es presentar un caso de displasia fibrosa poliostótica en la región craneofacial, al cual se le realizaron estudios de imagen, biopsia incisional para diagnóstico definitivo y remodelación ósea para mejorar el contorno facial y la calidad de vida del paciente.
Palabras clave: Displasia fibrosa monostótica, displasia fibrosa poliostótica, displasia fibrosa craneofacial, síndrome de McCune- Albright, síndrome de Jaffe-Lichtenstein
Advanced craniofacial fibrous dysplasia of bone remodeling
ABSTRACT
Fibrous dysplasia is an abnormal bone growth, it causes abnormal growth or swelling of bone maturation and differentiation. Fibrous dysplasia represents 2.5% of all bone tumors and more than 7% of benign tumors. The aim of this report is to present the findings of a polyostotic fibrous dysplasia case in the craniofacial skeleton. An incisional biopsy was done to make the diagnosis, after that, a bone remodeling surgery was performed to improve the facial contour and in general to improve the quality of life of the patient.
Key words: Monostotic fibrous dysplasia, polyostotic fibrous dysplasia, craniofacial fibrous dysplasia, McCune-Albright syndrome, Jaffe-Lichtenstein syndrome.
Recibido: Julio 2014 Aprobado: Noviembre 2014
INTRODUCCIÓN
El término displasia fibrosa fue propuesto por primera vez por Liechtenstein en 1938 para designar formas múltiples de osteítis fibrosa diseminada y esta condición fue descrita por Albrigth en 1931 (1). Chen y Noordhoff (1990) describen la displasia fibrosa como una lesión fibro-ósea benigna donde un tercio del tejido óseo normal es sustituido por tejido celular fibroso (2). Posteriormente en 1999, los autores coinciden con este criterio al plantear que la displasia fibrosa es una condición fibro-ósea benigna que consiste en la sustitución de tejido óseo por tejido fibroso (1).
La displasia fibrosa es una enfermedad de la maduración y remodelación ósea en la cual el hueso medular y cortical normal es reemplazado por hueso lamelar fibroso desorganizado. El tejido fibro-óseo resultante, es un hueso más elástico y estructuralmente más débil que el hueso original. Actualmente esta enfermedad resulta de un defecto en la maduración ósea que inicia en la histodiferenciación durante la fase embrionaria, una mutación o deleción genética que ocurre en el gen que codifica un transductor de proteína intracitoplasmática requerida para la maduración ósea. No hay evidencia que sugiera la presencia de displasia fibrosa por influencia hereditaria (3).
Histológicamente, la displasia fibrosa muestra trabéculas de hueso inmaduro en el seno de un estroma fibrocelular en el que se pueden encontrar osteoclastos dispersos. Las trabéculas son delgadas, no estando conectadas unas con otras y muestran una morfología curvilínea, con "patrón de letras chinas" (3). Radiográficamente se observa una imagen radiopaca discreta con aspecto de vidrio esmerilado, difusa, de límites mal definidos; puede ser unilocular o multilocular, y muchas veces está asociada con expansión de la cortical ósea. El mejor método para identificar la displasia fibrosa craneofacial es la tomografía computarizada, la cual permite determinar con exactitud la localización y extensión de la patología, además es un excelente auxiliar en el plan de tratamiento y en el procedimiento quirúrgico (4).
La displasia fibrosa representa alrededor del 2,5% de todos los tumores óseos y más del 7% de los tumores benignos. Los huesos craneales y faciales de los pacientes con displasia fibrosa se ven afectados en un 30% (7). La edad media de los pacientes con displasia fibrosa es de 25,8 años de edad, sin predilección por ningún género y se manifiesta antes de la tercera década de vida (3).
Se describen diferentes tipos de displasia fibrosa: la monostótica, la poliostótica y la craneofacial. La displasia fibrosa monostótica afecta a un solo hueso, mientras que la displasia fibrosa poliostótica afecta a dos o más huesos (Shafer 1987, Stewart 1989) (2); la displasia fibrosa craneofacial afecta huesos craneales y faciales. Una forma poliostótica acompañada de anormalidades endocrinas es conocida como síndrome de McCune-Albright o Jaffe Lichtenstein (5). El síndrome de McCune-Albright (MAS) es un trastorno sistémico raro caracterizado por la tríada de la displasia fibrosa poliostótica, trastornos endocrinos, manchas color café y pigmentación de la piel. El 90% de los pacientes con MAS presentan displasia fibrosa en el área craneofacial, causando una deformación significativa orofacial, trastornos dentales y dolor óseo (6).
La displasia fibrosa monostótica, representa alrededor del 75% de los casos, las zonas más comúnmente afectadas de los huesos faciales son el maxilar y la mandíbula (3). Además de los huesos faciales, otros huesos del cráneo como el esfenoides y el etmoides también pueden estar comprometidos; por lo que esta variedad ha sido llamada displasia fibrosa craneofacial (8, 9). Cuando el maxilar es afectado, el aumento de volumen es indoloro, unilateral, lento, progresivo y produce asimetría facial. Cuando se compromete el hueso temporal, se puede presentar pérdida de la audición; si el hueso frontal, el esfenoides o el etmoides son los huesos comprometidos, el resultado suele ser la obstrucción nasal y obliteración del ostium sinusal.(10).
El compromiso de la base del cráneo y de la órbita, en muchos casos produce diplopía, pérdida de la visión, parestesias en la región de distribución del nervio trigémino, cefalea y exoftalmia. Si el crecimiento óseo es rápido el resultado será sintomatología dolorosa (11).
La displasia fibrosa craneofacial es una forma muy poco frecuente, que se identifica por lesiones confinadas a huesos contiguos del esqueleto craneofacial, sin afectación de huesos extracraneales y sin alteraciones endocrinas, por lo que no se puede incluir en el resto de formas y es considerada como una entidad aparte. El 50% de las formas poliostóticas afectanla región craneofacial y hasta un 10% de las formas monostótica la presentan (12).
La etiología de la displasia fibrosa es probablemente, una mutación en el gen Gsa (GNAS1) del cromosoma 20q11. Esta mutación puede ocurrir durante el desarrollo embrionario o la vida postnatal. La mutación del gen Gsa produce un aumento de la adenilato ciclasa, que aumenta el AMPc intracelular, el cual es un nucleótido que funciona como segundo mensajero en varios procesos biológicos. La alta concentración del AMPc intracelular genera un aumento en la proliferación y diferenciación inapropiada de las células mutadas, causando la formación de una matriz fibrosa inmadura y desorganizada, generando el tejido fibroso de la displasia (13). Cuando el embrión se encuentra en la sexta semana del desarrollo, la mayoría de la histodiferenciación y migración de las células ya ha ocurrido. Si el mismo defecto genético se produce en esta época, la formación de células hijas se localiza en una región y por lo tanto puede producir el tipo de displasia fibrosa craneofacial, que involucra a varios huesos contiguos en un área determinada. Si el defecto genético se produce un poco más tarde, las células hijas afectadas serán menos y por lo tanto se produce la displasia fibrosa monostótica (3).
En consecuencia, todas las células hijas procedentes del aberrante celular original carecen de este transductor de señal, y por lo tanto una cierta población de células en el individuo será capaz de producir solamente hueso displásico fibroso en lugar de hueso maduro. Si el defecto genético se produce al principio del desarrollo embrionario, un gran número de células hijas se verán afectadas, algunos de las cuales aún no han migrado a su sitio del esqueleto eventual. Cuando las células migran en varios sitios del esqueleto, se produce la displasia fibrosa poliostótica. Si el defecto genético se produce al principio en el desarrollo embrionario, la célula original puede producir células hijas de diferenciación divergente. Si algunas células migran en el primordio óseo y algunos en la glándula endocrina primordial, producen ya sea el síndrome de McCune- Albright o síndrome de Jaffe-Lichtenstein de la displasia fibrosa poliostótica. Se cree que el momento en que estas alteraciones genéticas se producen es antes de la sexta semana de vida embrionaria (3).
A pesar de que el patrón anatomopatológico es característico, se debe realizar el diagnóstico diferencial con múltiples entidades como el fibroma osificante, el querubismo, el granuloma de células gigantes, el quiste óseo aneurismático, el cementoma gigante, el ameloblastoma o la displasia ósea, entre otros (3,12).
El fibroma osificante es una verdadera neoplasia consistente en una proliferación de un tejido fibroso celular, que aparece bien delimitada o raramente encapsulada, que incluye hueso neoformado o calcificaciones esféricas. El tumor solo se presenta en los maxilares y huesos craneofaciales, tiene su inicio en tercera y cuarta década de la vida. Histológicamente, el componente fibroso muestra fibroblastos que no presentan atipia, escasas células multinucleadas gigantes, y ocasionales macrófagos espumosos. Las trabéculas óseas son finas, curvilíneas y aparecen inconexas, no hallándose bordeadas por un ribete de osteoblastos. Las características histológicas son similares a las de la DF por lo que la distinción hay que hacerla en base a los criterios clínicos, radiológicos, hallazgos quirúrgicos y características (12).
El querubismo es una enfermedad benigna, autosómica dominante, que consiste en una lesión de células gigantes de la mandíbula/maxilar que se presenta en niños entre 2-5 años como una tumefacción bilateral indolora, con progresión hasta la pubertad, cuando regresa de manera espontánea (12).
Se ha reportado en la literatura, la transformación maligna de la displasia fibrosa a sarcoma en un porcentaje muy bajo de pacientes (0,4% en displasia fibrosa monostótica y hasta un 4% en McCune Albright). La región craneofacial es el lugar de malignización más frecuente con un 35,6% de los casos, en el fémur con una frecuencia del 24,7% y en la tibia con una frecuencia del 12,8% (14).
El propósito del presente trabajo es dar a conocer un caso de displasia fibrosa craneofacial, mencionando el manejo del paciente, tratamiento y seguimiento de dicha afectación.
DESCRIPCION DEL CASO
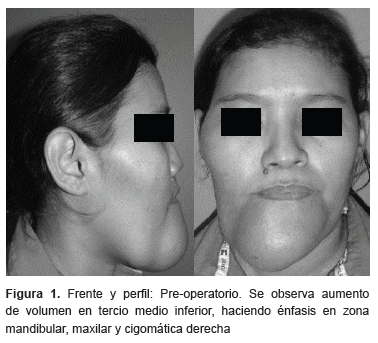
Paciente femenino de 45 años de edad quien acude a consulta por presentar aumento de volumen en la región facial de aproximadamente 20 años de evolución. A la exploración física se observa asimetría facial caracterizada por aumento de volumen en región mandibular, maxilar y cigomática derecha (figura 1). Actualmente acude refiriendo aumento de volumen progresivo asintomático en el tercio facial medio e inferior de predominio derecho, sin antecedentes sistémicos de importancia para el padecimiento actual, a la exploración física, observamos un cráneo normocefalico sin exostosis ni hundimientos, globos oculares simétricos con pupilas isocóricas y normoreflexicas a estímulos luminosos, aumento de tercio facial medio de predominio derecho, con desviación nasal a la izquierda, tercio facial inferior con aumento de volumen en región mentoniana y desviación mandibular a la derecha presentando tumor en región submandibular y submentoniano, perfil cóncavo, aumento de la distancia cervicomental, sin adenomegalias, intraoralmente se observa aumento de volumen en piso de boca, reborde alveolar anterior y lateral derecho, de consistencia dura, mucosa adyacente de color y características normales.
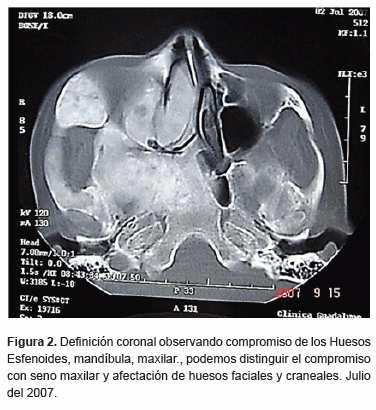
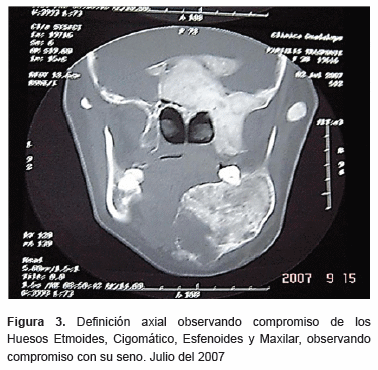
En estudios de imagen (tomografía axial computarizada) definiciones axiales y coronales (figura 2), (figura 3) se observa imagen mixta, con aspecto de vidrio esmerilado en algunas zonas y otras con áreas hipodensas y expansión de corticales, involucrando el hueso mandibular, maxilar, malar, el seno maxilar, región nasal del lado derecho, hueso etmoides y esfenoides. Se observa además desplazamiento órganos dentarios anteriores mandibulares.
Se decide realizar biopsia incisional con diagnóstico definitivo, reportando displasia fibrosa la cual confirma nuestro diagnóstico de displasia fibrosa craneofacial, se decide hacer remodelación ósea con fines estéticos y funcionales, bajo anestesia general balanceada e intubación nasotraqueal, se realiza infiltración con lidocaína al 2% con epinefrina 1:100.000 con fines hemostáticos, se procede a realizar abordaje submental (extraoral) a través de cicatriz previa, se diseca por planos y se expone región submandibular y mentoniana, se realiza remodelación y toma de muestra de la zona hasta obtener adecuado contorno, se sutura por planos, se verifica hemostasia y se coloca cura compresiva, seguidamente se procede a realizar abordaje circunvestibular maxilar derecho se levanta colgajo mucopériostico y se expone zona de displasia en región cigomático maxilar derecha, se realiza remodelación ósea y toma de muestra de la zona hasta lograr adecuado contorno, se sutura por planos y se verifica hemostasia, y se termina el procedimiento sin complicaciones. Se envía especímenes para estudios histopatológicos reportando hueso inmaduro en el seno de un estroma fibrocelular en el cual se pueden encontrar osteoclastos dispersos, trabeculado delgado e irregular, no estando conectados unos con otros mostrando morfología curvilínea (patrón de letras chinas). Se solicita interconsulta con endocrinología quienes descartan compromiso sistémico del padecimiento. Se realiza seguimiento a la paciente durante 5 años observando evolución satisfactoria y adecuada estética. (figura 4), (figura 5).


DISCUSIÓN
Una vez realizado el diagnóstico definitivo a través de estudios histopatológicos de la displasia fibrosa, es necesaria la interconsulta para descartar una alteración endocrina.A través de estudios de imagen general se puede descartar extensión extracraneal de la displasia fibrosa (radiografías simples, tomografías o gammagrafías óseas). Algunos autores consideran que la medición de la fosfatasa alcalina sérica (FAS), junto con la edad del paciente, pueden ser un buen marcador para detectar la progresión de la displasia fibrosa craneofacial (15).
Respecto al tratamiento médico, históricamente la radioterapia ha sido una de las opciones a considerar, pero actualmente ha sido abandonada por el riesgo de transformación maligna (sarcoma post radioterapia). La quimioterapia ha demostrado no ser efectiva en estas afecciones (14). Actualmente se plantea la terapia con bifosfonatos, con la coadyuvancia del calcio, la vitamina D y el fósforo (16), sin embargo existen contradicciones respecto a esta opción terapéutica. Autores como Plotkin,no han demostrado evidencia de mejoría radiológica en sus estudios, a pesar de haber obtenido resultados estadísticamente significativos respecto a los niveles de los marcadores óseos (17). Algunos reportes concluyen que la terapia intravenosa con bifosfonatos es segura, incluso para tratamientos a largo plazo. A pesar de todos los esfuerzos, con el uso de bifosfonatos no se han logrado resultados efectivos para la displasia fibrosa, porque no revierten la mutación de las células osteoblásticas, aunque sí son una terapia alternativa no quirúrgica al tratamiento, porque inducen mejoría radiológica y bioquímica, además de actuar sobre el dolor de origen óseo. Sin embargo, son necesarios estudios adicionales para establecer plenamente la eficacia del uso de bifosfonatos en la displasia fibrosa (12).
La remodelación ósea, la excisión radical y reconstrucción (propuesta por primera vez en 1990 por Chen y Noorhoff), son los tratamientos más aplicados en la actualidad en los casos de displasia fibrosa. Las indicaciones de dichos procedimientos quirúrgicos engloban las alteraciones funcionales y estéticas de la zona afectada, entre las cuales se incluye la deformidad estética, dolor, disminución de la audición, obstrucción nasal o sinusal, fractura en sitio de patología y la compresión del nervio óptico (18).
La cirugía microvascular es considerada una opción de tratamiento. El injerto libre de peroné fue la cirugía propuesta por Taylor y colaboradores en 1977 para el tratamiento de la displasia fibrosa (19), posteriormente esta técnica fue modificada por Hidalgo en 1989 (20) y es actualmente la técnica para la reconstrucción microvascular de mandíbula, posterior a resecciones quirúrgicas, ya que permite una longitud ósea aceptable con un excelente aporte vascular.
La remodelación ósea es la técnica de elección para muchos autores en nuestro caso fue el tratamiento de
elección. Es necesario destacar que en cada caso es preciso individualizar el tratamiento, y tener en cuenta que en muchas ocasiones es difícil la excisión completa debido a su localización, habiendo un gran porcentaje de recidivas (10 a 25%), lo que obliga un seguimiento continuo de estos pacientes.
CONCLUSIÓN
La displasia fibrosa es una patología de poca incidencia que se presenta en las primeras tres décadas de la vida. La forma monostótica es más común que la poliostótica y una variedad peculiar de la poliostótica es el Síndrome de McCune-Albright.
Numerosos reportes indican que es resultado de una anormalidad del mesénquima osteógeno durante el desarrollo. El trastorno no es hereditario. Para llegar a un diagnóstico correcto, son muy importantes tanto las técnicas de imagen como el análisis histopatológico, pues permite hacer el diagnóstico diferencial con otras lesiones como el fibroma osificante o con la enfermedad de Paget.
Imagenologicamente pueden observarse imágenes mixtas (radiolúcidas y radiopacas) de acuerdo al grado de formación de tejidos calcificados he aquí la importancia del diagnóstico radiológico que podamos hacer con nuestros pacientes día a día.
Es una lesión ósea benigna, evoluciona lentamente y afecta con mayor frecuencia los maxilares, causa deformidad ósea, afecta la estética facial y la autoestima del paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Sávio de Souza E, Cristina da Costa M, Bezarra L, Pereira L, Batista de Souza L. Displasia Fibrosa: Relato de Caso Clínico. Revista Brasileña de Patología Oral 2006.
[ Links ]2. Saueressig F, Gerhardt de Oliveira M. Displasia Fibrosa Poliostótica asociada a Síndrome de McCune –Albright: Relato de caso. Revista Brasileña de Patología Oral 2004.
3. Robert E Marx, Stern D. A rationale for Diagnosis and treatment. Oral and Maxillofacial Pathology; 2003. Pag 747-754.
4. Reed RJ. Observations of bone maturation and fibrous
dysplasia of bone. Bull Tulane Med Fac. 1959;19:45–53. [ Links ]5. Martin Etchart. Capítulo 12. Anatomía Patológica Osteoarticular.
Pontificia Universidad Católica de Chile. Escuela de Medicina. Último acceso 25 de junio, 2008.6. Sunday O. Akintoye, Alison M. Boyce, Michael T. Collins.
Dental perspectives in fibrous dysplasia and McCune–Albright syndrome. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology 2013:116:e149-e155.7. Pinsolle V, Rivel J, Michelet V, Majoufre C, Pinsolle J. Treatment
of fibrous dysplasia of the cranio-facial bones. Report of 25 cases. Ann Chir Plast Esthet. 1998; 43:234-239. [ Links ]8. Regezi JA, Sciubba JJ. Patologia bucal: correlações clinicopatológicas. 3 ed. Philadelphia, Guanabara-Koogan; 2000. p.324-326.
[ Links ]9. Ricalde P, Horswell B. Craniofacial fibrous dysplasia of the fronto-orbital region: A case series and literature review. J Oral Maxillofac Surg 2001; 59:157-168. [ Links ]
10. Crawford LB. An unusual case of fibrous dysplasia of the maxillary sinus. Am J Orthodontics Dentofac Orthoped 2003; 124-126. [ Links ] 11. Michael CP, Lee AG, Patrinely JR, Stal S, Blacklock JB. Visual 12. Ventura-Martínez N, et al. Displasia Fibrosa craneofacial avanzada: a propósito de un caso. Rev Esp Cir Oral Maxilofacial 2012; 04: 002. [ Links ] 13. Perdigao PF, Pimenta FJ, Castro WH, De Marco L, Gomez 14. Kim D, Ghali GE, Wright JM. Surgical treatment of giant 15. Park BY, Cheon YW, Kim YO. Prognosis for craniofacial dysplasia after incomplete resection: age and serum alkaline phosphatase. Int J Oral Maxillofac Surg. 2010; 39:221-226.
16. Chapurlat RD. Medical therapy in adults with fibrous dysplasia of bone. J Bone Miner Res. 2007;21:114-119. [ Links ] 17. Plotkin H, Rauch F, Zeitlin L. Effect of pamidronate treatment 18. Chen YR, Noordhoff MS. Treatment of craniomaxillofacial 19. Ventura-Martínez Natalia, Guijarro-Martínez Raquel, Morales-Navarro Juan Diego, Solís-García Ignacio, Puche-Torres
20. Hidalgo D. Fibular free flap: a new method of mandible reconstruction. Plast Reconstr Surg. 1989;84:71-págs. [ Links ]
