










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Puericultura y Pediatría
versión impresa ISSN 0004-0649
Arch Venez Puer Ped v.71 n.4 Caracas dic. 2008
Encefalomielitis aguda diseminada
María Elena Ravelo (*), Norelis Rodríguez (**), Mariana Jiménez (***), Teresa Espinette (**), Manuel González (***), Ángel Sánchez (**).
(*) Jefe de Servicio de Neurología,
(**) Adjuntos del Servicio de Neurología,
(***) Residentes del Servicio de Neurología. Hospital J. M. de los Ríos. Caracas-Venezuela
Autor corresponsal: Dra. María Elena Ravelo Jefe del Servicio de Neurología Hospital J. M. de los Ríos.Av. Vollmer, San Bernardino Caracas-Venezuela Correo electrónico: meravelo@gmail.com
RESUMEN
La encefalomielitis aguda diseminada (EMAD) es la leucoencefalopatía adquirida de mayor presentación en la edad pediátrica, de naturaleza inflamatoria-autoinmune, generalmente monofásica, polisintomática, y asociada a compromiso del estado de conciencia.
Frecuentemente es precedida por un proceso infeccioso o por una inmunización. Se revisó la literatura médica de los últimos años considerando los aspectos etiopatogénicos, clínicos, diagnósticos y terapéuticos. Se presentan los últimos criterios diagnósticos para la EMAD y sus formas recurrentes, así como la importancia de los diagnósticos diferenciales, entre ellos la Esclerosis Múltiple Pediátrica (EMP). Las imágenes de Resonancia Magnética (IRM) constituyen el estudio de elección para detectar lesiones desmielinizantes.
Palabras Clave: encefalomielitis aguda diseminada, complejo trimolecular, autoinmune desmielinización, resonancia magnética, esclerosis múltiple pediátrica, esteroides.
SUMMARY
Acute disseminated encephalomyelitis (ADEM) is an acquired leucoencephalopathy, that occurs more frequently during childhood, of inflammatory autoimmune condition, usually monophasic, polysymptomatic and associated with sensory deterioration. It is a frequently preceded by infections or vaccinations. Recent medical literature was reviewed considering etiology, pathogenesis, clinical, diagnosis and treatment aspects. The current diagnostic criteria for ADEM, its variants and the relevance of differential diagnostics, like Pediatric Multiple Sclerosis (PMS) are included. Magnetic Resonance imaging (MRI) is recommended to detect demyelination injuries. High doses of steroids are still the therapeutic alternative for ADEM.
Key Words: Acute disseminated encephalomyelitis, trimolecular complex, autoimmune, demyelination, magnetic resonance imaging, diagnostics criteria, pediatric multiple sclerosis, steroids.
INTRODUCCIÓN:
La EMAD es una enfermedad inflamatoria de naturaleza autoinmune que compromete predominantemente la sustancia blanca del SNC y cuya lesión fundamental es la desmielinización. Habitualmente está precedida por procesos infecciosos (virales, bacterianos, parasitarios) o la colocación de vacunas; sin embargo, se han descrito otras situaciones tales como la administración de sueros y fármacos (sulfas, antiparasitarios); de igual manera se han reportado pacientes sin una causa previa aparente (1, 2). Por este motivo se han utilizado términos como encefalomielitis postinfecciosa o parainfecciosa dependiendo del contexto de la historia clínica del paciente (1,3,4).
Dentro de las enfermedades desmielinizantes de la infancia, la EMAD es la más frecuente (5-7). Clínicamente se caracteriza por la aparición aguda de manifestaciones neurológicas polisintomáticas y generalmente asociada a compromiso del estado de conciencia, relacionándose con la afectación central de tipo multifocal (1,4,5,8).
El curso clínico es generalmente monofásico, aunque se describen casos de recidivas o multifásicos, constituyendo todo un reto para el diagnóstico diferencial con Esclerosis Múltiple Pediátrica (1,2,4,6,9-11).
Debido a la falta de marcadores biológicos específicos (11-14), el diagnóstico estará basado en la clínica general y neurológica del paciente: forma de inicio, edad de presentación, evolución, signos y síntomas acompañantes y estudios complementarios de soporte, como el estudio del líquido cefalorraquídeo (LCR) y la Resonancia Magnética (RMN) (1,5,6,9,12).
La descripción histopatológica está relacionada con una mielinoclasia de ubicación peri-vascular que produce numerosos focos de desmielinización perivenosa con preservación, generalmente, de los axones. Se evidencian infiltrados de células mononucleares que se disponen alrededor de venas y vénulas, así como células microgliales y macrófagos en las áreas de desmielinización (3,4,6).
El mecanismo inmunopatológico está basado en una respuesta de tipo autoinmune por antígenos autólogos (6,15,16), inducida generalmente por agentes infecciosos o vacunas. Se plantea un mecanismo inflamatorio autoinmune, similar al de la encefalomielitis alérgica experimental, la cual se produce por virus murino Theiler en ratas susceptibles, produciendo encefalitis aguda y desmielinización (6,10,17-19), relacionado con la producción de anticuerpos policlonales contra el agente etiológico y contra diversas estructuras del SNC del huésped, principalmente contra la mielina. Se postula que se debe a una reacción cruzada entre la respuesta inmune celular y humoral contra antígenos virales y bacterianos y antígenos de mielina, lo que lleva a una desmielinización mediada por complejos inmunes (6,10).
La afectación neurológica descrita en la EMAD incluye alteración del nivel de conciencia, ataxia, déficit motor focal, cefalea, afasia, convulsiones, alteración del control de esfínteres, neuritis óptica o afectación de otros pares craneales, entre otras que traducen la afectación multifocal del SNC (1,7,8).
El diagnóstico se basa en la correlación de la clínica con los hallazgos en la RMN que muestra lesiones desmielinizantes, hiperintensas en T2 y FLAIR, en el mismo estadío evolutivo, con frecuencia bilateral de predominio en la sustancia blanca hemisférica, aunque la sustancia gris también puede estar afectada (1,7,12,20,21).
El pronóstico de la EMAD es generalmente favorable y la mejoría se produce con la administración de dosis altas de esteroides (metlprednisolona), lo cual puede acortar la duración de los síntomas neurológicos y detener su progresión o secuelas (1-3,5,7,14). La mortalidad en niños, que alcanzaba el 25% hace tres décadas ahora es inferior al 10% (6,14,20).
EPIDEMIOLOGÍA:
La incidencia de EMAD en nuestro país es desconocida. Leake et al (8) establecieron una incidencia de 10,4/100.000 por año en una población de Estados Unidos, cuadriplicando el valor en los últimos años de su estudio (1998-2000), debido probablemente al uso extendido de la RMN (8,10). No hay un claro predominio del sexo en la EMAD, sin embargo hay varios reportes de una mayor frecuencia en varones (relación varón: hembra de 1,3:1 (2,7,8) a 1,8:1 (21).
La edad promedio de presentación se encuentra entre los 5 y los 8 años (1,20,21-23). Se ha descrito que es más frecuente en la raza blanca que en la negra 6:1 (2,7,8) y que tiene cierta relación con el período estacional, presentándose generalmente en invierno o primavera (1,4,9).
ETIOPATOGENIA:
Cerca del 75% de los niños con EMAD tiene antecedentes de cuadro febril previo (1,3,8,9), pero el porcentaje de confirmación del agente asociado a EMAD es bajo y usualmente no supera el 17-24% (14).
Dentro de los agentes etiológicos, el virus del sarampión constituye el principal agente y responsable de los casos más graves (1,6). La probabilidad de presentar EMAD es de 1:1000 para el virus del sarampión, en cambio es mucho más baja para una infección por varicela o rubéola. Otros virus asociados son herpes simple I-II, gripe, Epstein Barr, herpes humano tipo 6, citomegalovirus, paratoditis, Coxsackie, hepatitis A, dengue, arbovirus, retrovirus, polio (2,6,12,24-30). Entre los agentes no virales están Borrelia, Micoplasma pneumoniae, estreptococo beta hemolítico, Campilobacter y Plasmodium (3,6,7,31,32).
Se han descrito un número importante de casos relacionados con las inmunizaciones. Los primeros casos descritos se relacionaron con la vacuna antirrábica (Semple). En la actualidad, la EMAD se relacionan principalmente con las vacunas antisarampión, gripe, parotiditis, pertussis, polio (1,33).
La patogenia de la EMAD, según estudios realizados en animales, parece ser mixta: se suman mecanismos inflamatorios y de respuesta autoinmunitaria de tipo celular y humoral que conllevan a la destrucción de la mielina, y por ende a la desmielinización del SNC (1,4,6,7).
En la generación de la cascada autoinmunitaria, que culmina con la destrucción de la mielina del SNC, participan linfocitos T del tipo CD4 y CD8 (15,35). Las células T son activadas a través de la presentación del antígeno; las Células Presentadoras de Antígeno –APC- (células dendríticas, macrófagos, microglia, etc) procesan la molécula, y parte de ésta (X) es presentada en la superficie celular unida a la molécula del Complejo Mayor de Histocompatibilidad (MHC) clase II. La combinación MHC/X es reconocida por el Receptor específico de la Célula T (TCR). El complejo TCR y MHC/X es conocido como complejo trimolecular y es la primera señal en la presentación antigénica; sin embargo, para una activación exitosa de la célula T es necesaria una segunda señal, proporcionada por las moléculas co-estimulantes (ej. CD28 sobre la célula T y B7 sobre la APC) (Figura 1) (34). El complejo trimolecular constituye la base común para la activación de la respuesta inmune; sin embargo, se realizan numerosas investigaciones con la finalidad de determinar si existen características propias para este complejo, en ciertas entidades desmielinizantes, como por ejemplo en EMAD o EMP. En EMAD el complejo mayor de histocompatibilidad se ha asociado con alelos clase II HLA-DRB1, HLA-DRB3, HLA-DRB5 (1).

Todos los individuos normales tienen grupos de células T virgen cuyos Receptores de Células T (TCR) están programados durante el desarrollo para un Complejo Mayor de Histocompatibilidad (MHC)/antígeno determinado. Luego de la activación de la célula T, se produce una expansión clonal y las progenies se diferencian en subtipos Th1 y Th2, los cuales tienen una función efectora específica en el sistema inmunológico, ejemplo: activación de macrófagos por Th1, estimulación de linfocitos B por Th2 (Figura 2) (34).

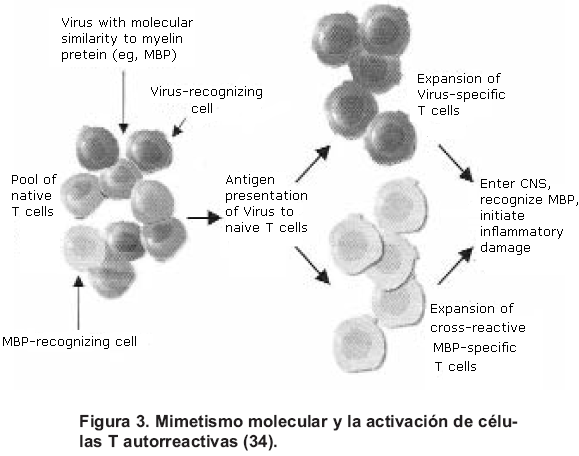
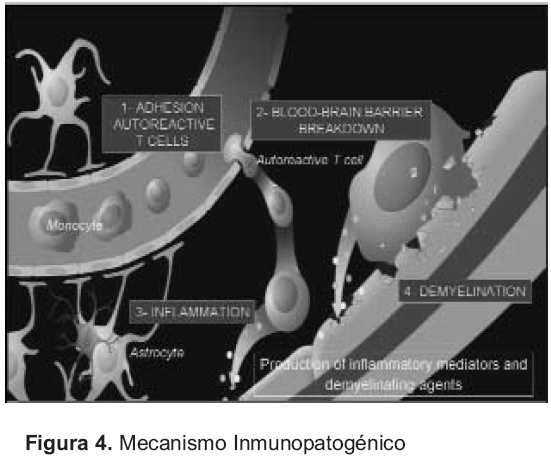
La activación de células T autorreactivas es el paso clave en la autoinmunidad; hay varias vías que conducen a la activación de células T autorreactivas, y una de ellas es el mimetismo molecular por el que agentes (ej. virus) similares a proteínas endógenas (ej. proteína básica de la mielina –MPB-) son presentadas a las células T; esto lleva a la activación de células T virus-específicas, así como de células T MPB-específicas. Ambas poblaciones activadas se adhieren a los vasos sanguíneos, atraviesan la barrera hematoencefálica y migran hacia el interior del SNC, facilitando luego la entrada de otros linfocitos y macrófagos, que ocasionan la inflamación y la desmielinización (Figuras 3 y 4) (34).
La histopatología de la EMAD muestra áreas de inflamación y desmielinización perivenosas, con infiltración de monocitos, neutrófilos y macrófagos, cargados de lípidos; en el estadio tardío se evidencia hiperplasia astrocítica y gliosis (15,16,35,36) (Figura 5).

CLÍNICA:
El curso es generalmente monofásico, presentándose 2 a 4 semanas después del estímulo antigénico. Como se mencionó anteriormente, aproximadamente alrededor del 70% de los pacientes reportan antecedente infeccioso o de vacunación previo a la clínica, aunque existen algunos casos sin antecedente demostrable (2,12).
Los síntomas o signos clínicos típicos pueden aparecer en una forma abrupta, con una rápida progresión a encefalopatía aguda o puede estar precedido de una fase prodrómica con fiebre, malestar, cefalea, náuseas o vómitos antes de las manifestaciones clínicas del EMAD. El curso por lo general es rápidamente progresivo, observándose máximo déficit neurológico en 4 a 5 días (1,2,7,9).
En diferentes series de trabajos sobre EMAD, la sintomatología neurológica descrita incluye signos piramidales unilaterales o bilaterales (60 - 95%), hemiplejia aguda (76%), ataxia (18 a 65%) parálisis de los nervios craneales (22-45%) afectación visual o neuritis óptica (7 a 23%), convulsiones (13 a 35%), compromiso de la médula espinal (24%), afectación del habla (5 a 21%), hemiparestesias (2-3%), con afectación demostrada del estado mental o de la conciencia, llegando al estupor o coma (1,2,9,12,21). En uno de los estudios publicados se describen pacientes (lactantes) con crisis epilépticas focales motoras persistentes derivando a status epiléptico (21,37).
En los pacientes con manifestaciones clínicas compatibles con EMAD y signos de afectación medular (mielitis) se deberá contemplar la afectación de dicha zona. (7,12). De igual manera, el compromiso nervioso periférico o polirradículoneuritis, deberá diferenciarse del Síndrome de Guillan-Barré observando si existe compromiso encefálico (20,38).
El Grupo de Trabajo Internacional para el estudio de EMP y Desórdenes Relacionados (como la EMAD) propuso en el 2007 una serie de definiciones, tanto clínicas como radiológicas, para incluir a los pacientes en cada categoría de afectación de la sustancia blanca o desmielinización (Figura 6) (1,22).

EMAD MONOFÁSICA:
1. Paciente con un primer evento clínico de causa presuntamente inflamatoria o desmielinizante, de inicio agudo o subagudo que afecte de manera multifocal áreas del SNC. La presentación clínica debe ser polisintomática y debe incluir encefalopatía, dada por uno o más de los siguientes: a. Cambios del comportamiento, por ejemplo: confusión, irritabilidad excesiva. b. Alteración del estado de conciencia, por ejemplo: letargia, coma.
2. El evento deberá ser seguido de mejoría tanto clínica como por RMN, aunque puede haber déficit residual.
3. El paciente no tiene historia de episodios clínicos con hallazgos de eventos desmielinizantes previos.
4. Ninguna otra etiología puede explicar el evento.
5. Los síntomas o signos clínicos nuevos o fluctuantes, o los hallazgos de la RMN que ocurran en los 3 meses siguientes al inicio de la EMAD, son considerados parte del evento agudo.
6. Las imágenes por RMN muestran lesión(es) focal o multifocales que involucran predominantemente la sustancia blanca, sin evidencia radiológica previa de cambios destructivos de la sustancia blanca.
a. RMN cerebral con imágenes ponderadas en T2 y FLAIR revela lesiones grandes (> 1 ó 2 cm), multifocales, hiperintensas y localizadas en la sustancia blanca de las regiones supratentoriales o infratentoriales; el compromiso de la sustancia gris, especialmente ganglios basales y tálamo, es frecuente.
b. En pocos casos las imágenes de RMN cerebrales muestran una lesión única grande (≥ de 1 a 2 cm) que afecta predominantemente a la sustancia blanca.
c. La RMN a nivel medular muestra lesión(es) confluentes intramedulares con realce variable, adicional a una RMN cerebral anormal con los hallazgos previamente especificados.
EMAD RECURRENTE:
1. Nuevo evento de EMAD con recurrencia de los síntomas y signos neurológicos iniciales, 3 ó más meses después del primer evento, sin que se vean involucradas nuevas áreas clínicas por historia, examen físico o neuroimagen.
2. El evento no ocurre mientras el paciente recibe esteroides, y se presenta al menos 1 mes después de culminada la terapia esteroidea.
3. La RMN no muestra nuevas lesiones; las lesiones iniciales pueden observarse agrandadas.
4. No existe una mejor explicación para el evento.
EMAD MULTIFÁSICA:
1. Evento de EMAD seguido de un nuevo evento que también encuentra criterios para EMAD, pero que involucra nuevas áreas anatómicas del SNC confirmadas por historia clínica, examen neurológico y neuroimagen.
2. El nuevo evento debe ocurrir al menos 3 meses después del inicio del primer evento EMAD y al menos 1 mes después de haber completado la terapia esteroidea.
3. El nuevo evento de EMAD debe mostrar una presentación polisintomática incluyendo encefalopatía, con signos y síntomas neurológicos que difieran del evento inicial (los cambios del estado mental pueden no diferir del primer evento).
4. La RMN cerebral debe mostrar nuevas áreas involucradas, pero también debe mostrar resolución parcial o total de las lesiones asociadas al primer evento de EMAD.
Diversos trabajos de EMAD describen que las recaídas representan el 5 al 21% (7-9,28,29); sin embargo, los criterios diagnósticos utilizados para definir fueron muy variados entre los estudios, lo cual explica la variabilidad en el porcentaje.
Encefalomielitis Diseminada (EMD), y dependiendo de la presentación clínica y radiológica, ubicarla en la clasificación propuesta anteriormente (39).
ESTUDIOS PARACLÍNICOS ESTUDIOS DE NEUROIMÁGENES:
El examen de elección para el diagnóstico y para ubicar las áreas de desmielinización del SNC es la RMN cerebral, ya que es altamente sensible en la detección de alteraciones de la sustancia blanca; en las secuencias T2 y FLAIR es posible visualizar prácticamente todas las lesiones (1-10,12); éstas son bilaterales, generalmente asimétricas y altamente variables en tamaño y número. Frecuentemente son de ubicación subcortical o sustancia blanca central, sustancia blanca periventricular, cerebelo, tallo encefálico y médula espinal (1-10,12,21); la sustancia gris profunda de tálamo y ganglios basales están frecuentemente comprometidas, generalmente en forma simétrica (21,23,39-43).
La afectación del cuerpo calloso es de baja frecuencia, sin embargo en lesiones extensas de sustancia blanca cercanas al cuerpo calloso pueden extenderse a través de éste y llegar al hemisferio contralateral (21,42).
Han sido propuestos cuatro patrones de compromiso del SNC para describir los diferentes tipos de lesiones evidenciadas en la RMN cerebral en las EMAD (21):
1. EMAD con lesiones pequeñas.
2. EMAD con lesiones grandes, confluyentes, tumefactivas con edema perilesional y efecto de masa.
3. EMAD con compromiso talámico bilateral y simétrico.
4. Encefalomielitis Hemorrágica Aguda asociadas a extensas áreas de desmielinización.
Igualmente, se han descrito áreas de compromiso en la médula espinal en la EMAD en 11-28% (44-46). La típica lesión medular es de tamaño significativo con tendencia a incrementarse, mostrando realce variable, y la región que generalmente se afecta es la torácica (46).
Con respecto a la relación temporal entre la clínica y la aparición de las alteraciones en la RMN cerebral, se ha demostrado que puede existir un desfase entre 2 a 25 días entre el inicio de los síntomas y la detección de las lesiones en la RMN (1,20,46- 49). Este hecho demuestra que una RMN normal al inicio del cuadro clínico no descarta la EMAD, y que todo niño con sintomatología neurológica polisintomática y persistente debe someterse a una nueva RMN cerebral y medular de 1 a 3 semanas después, para detectar posibles lesiones de aparición mas tardía (20).
La completa resolución de las anormalidades encontradas en la RMN después del tratamiento ha sido descrita en 37 a 75% de los pacientes con EMAD, y la resolución parcial en 25 a 53% de los pacientes (21,50), en un lapso de 6 meses.
No hay un criterio claro en cuanto al tiempo en que se debería realizar una RMN de control en los pacientes con EMAD, sin embargo los diferentes autores sugieren al menos dos estudios adicionales de neuroimagen por RMN después de la primera RMN normal, sobre un período de 5 años del evento inicial como la conducta más apropiada para asegurar la ausencia de lesiones (20,21,46-48) (Figuras 7 y 8).


Laboratorio y estudios neurofisiológicos: Los estudios de laboratorio de EMAD muestran frecuentemente elevación de los marcadores inflamatorios (leucocitosis, aumento de la velocidad de sedimentación). El estudio del líquido cefalorraquídeo puede ser normal o mostrar alteraciones inespecíficas en el 20 a 80% de los casos, con elevación de las proteínas y pleocitosis a expensas de mononucleares (1-4,20,21,42).
Uno de los hallazgos que mejor discrimina entre la Esclerosis Múltiple Pediátrica (EMP) es la presencia de síntesis intratecal de bandas oligoclonales (BOC) de IgG, que no se observan en el suero. Ocurre en el 40 a 95% de los pacientes con EMP y sólo en el 0 a 29% de niños con EMAD (1-4,7,9); es decir, la presencia de BOC de IgG son generalmente negativas en la EMAD (1,2,4,5,9,12,21), y por lo general en los pacientes con BOC positivas se observa negativización meses después (49-52) (a diferencia de lo que ocurre con las EMP, que son permanentes en el tiempo). Igualmente considerar que existen una serie de entidades o enfermedades que presentan BOC/Índice IgG positivas (Cuadro 1).
Cuadro 1. Enfermedades que cursan con BOC positiva (tomado de 53)
| Enfermedades Inflamatorias Esclerosis múltiple Lupus eritematoso sistémico Enfermedad de Behcet Síndrome de Sjogren primario Enfermedades Infecciosas Encefalitis viral Neuroborreliosis Neurosifilis Panencefalitis esclerosante subaguda Panencefalitis progresiva por rubéola Criptococosis de SNC Síndrome de Guillain-Barré y otras neuropatías periféricas Neoplasias y Síndromes paraneoplásticos Linfomas Lesiones expansivas Carsinomatosis meníngea Sarcoidosis |
BOC: bandas oligocionales
Los estudios electrofisiológicos ayudan al diagnóstico de EMAD. Los potenciales evocados somatosensoriales (PESS) permiten establecer la afectación medular. Los PESS y los potenciales evocados auditivos (PEA) detectan disfunción del tallo cerebral. Igualmente, los potenciales evocados visuales (PEV) son una herramienta valiosa para confirmar la afectación de la vía visual aferente, en especial en la neuritis óptica, que es generalmente unilateral en la EMP y con tendencia a compromiso bilateral en la EMAD (37,50,54). El electroencefalograma (EEG) muestra frecuentemente lentificación difusa del ritmo de base en la EMAD (2,4-6,21,42). En el 2% de los niños con EMAD se observa actividad epileptiforme (21).
DIAGNÓSTICOS DIFERENCIALES
Las encefalopatías agudas y los eventos desmielinizantes del SNC en niños representan un reto para pediatras y neuropediatras, ya que muchos procesos inflamatorios y no inflamatorios se puede presentar una clínica y hallazgos de neuroimagen similares, lo que hace mas difícil el diagnóstico. Ante un paciente con una encefalopatía aguda, en base a la historia clínica y al examen neurológico, la orientación inicial sería evaluar la presencia de un proceso infeccioso de naturaleza viral o bacteriana, ante la pronta necesidad de instaurar un tratamiento. La realización de la RMN cerebral y medular con técnica T2 y FLAIR y la administración de contraste paramagnético, conjuntamente con la realización de la punción lumbar y el estudio del líquido cefalorraquídeo permitiría una aproximación adecuada del diagnóstico, sea éste de naturaleza infecciosa como encefalitis o meningitis o de otra etiología (2,20,21).
La evidencia de un proceso inflamatorio expresado en el LCR por pleocitosis, elevación de proteínas y del índice de inmunoglobulinas, con demostración de la presencia del agente, sea éste viral, bacteriano o micótico, conduce a pensar en un proceso de naturaleza infecciosa.
Al no existir una clara evidencia de infección, el hallazgo de lesiones en la RMN cerebral, que comprometen predominantemente la sustancia blanca nos orienta hacia otras posibilidades diagnósticas como la EMAD, sin embargo otros diagnósticos diferenciales deben ser contemplados, como las vasculitis primarias o secundarias del SNC, leucoencefalopatía multifocal progresiva, neurosarcoidiosis, panencefalitis esclerosante subaguda, leucodistrofias, encefalopatías mitocondriales, leucoencefalopatías secundarias a quimioterapia o radioterapia, la mielinolisis osmótica y condiciones desmielinizantes relacionadas como la EMP (1,55-59), deberán ser considerados. (Cuadro 2). Para la EMP resulta útil utilizar los criterios establecidos por el Grupo Internacional de Estudio de EMP y Trastornos Relacionados (22), los cuales consideran:
La EMP requiere de múltiples episodios de desmielinización del SNC separados en tiempo y espacio, como especificado en adultos, eliminando cualquier límite inferior de edad (se incluye a los menores de 10 años).
La RMN puede ser usada para satisfacer los criterios de diseminación en el espacio requerida si se aplican los Criterios de McDonald; para una RMN positiva el estudio deberá mostrar 3 de estas 4 características: 1) >9 lesiones de materia blanca o 1 lesión realzada por Gadolinio, 2) ≥ 3 lesiones periventriculares, 3) 1 lesión yuxtacortical, 4) 1 lesión infratentorial.
La combinación de LCR anormal y 2 lesiones por RMN, de las cuales una debe estar en el cerebro, puede también satisfacer el criterio de diseminación en el espacio; el LCR debe mostrar tanto bandas oligoclonales como un índice elevado de IgG.
La RMN puede ser usada para satisfacer los criterios de diseminación en el tiempo después del evento clínico inicial, aun en ausencia de un nuevo evento desmielinizante; lesiones nuevas en T2 o realzadas con Gadolinio deben desarrollarse en los 3 meses siguientes al evento clínico inicial.
Un episodio consistente con las características clínicas de EMAD no puede ser considerado como un primer evento en Esclerosis Múltiple, a menos que el curso de la enfermedad siga las características ya descritas.
Cuadro 2 Diagnóstico Diferencial de la EMAD (en la columna de la derecha se mencionan algunas pruebas complementarias que pueden ser de utilidad (53).
| Diagnóstico diferencial Infecciones aguadas del SNC | Pruebas complementarias
|
| - Meningitis bacteriana | LCR, cultivo |
| - Mycoplasma | LCR, (PCR), serología, cultivo |
| - Listeria | LCR, cultivo |
| - Encefalitis viral: grupo herpes, rubéola, sarampión, VIH | LCR, (PCR), serología |
| - Hongos: toxoplasma | Neuroimagen |
| - Infecciones subagudas crónicas de SNC | |
| - Meningitis tuberculosa | LCR, ADA, Mantoux, Rx tórax, Cultivo |
| - Neurosifilis | LCR, serología |
| - Enfermedad del Lyme | LCR, serología |
| - Neurobrucelosis | LCR, serología, cultivo |
| - Síndrome de Reye | LCR, hiperamoniemia |
| - Vasculitis del SNC | Multisistémica. Curso subagudo-Crónico |
| - Panarteritis nodosa | ANCAp |
| - Chung Strauss | ANCAp |
| - Wegener | ANCAp |
| - Bechet | HLA B5 |
| - Lupus eritematoso | ANA |
| Sjögren | Biopsia glanular |
| Enfermedades glanulomatosas Sarcoidosis | Multisistématica. Curso subagudo-Crónico |
| Esclesosis múltiple y Variantes | Niveles de ECA |
| Evolución em brotes. LCR (bandas oligocionales). RM | |
| Otros | |
| Neoplasias, abscessos, trobosis venosa | Neuroimagen (RM, anglo RM). IG G y M |
Entre otras consideraciones, la presencia de lesiones en la RMN cerebral de tipo anular en la sustancia blanca es inusual en EMAD, por lo cual deberá excluirse abscesos cerebrales, tuberculomas, neurocisticercosis, toxoplasmosis e histoplasmosis (42).
TRATAMIENTO Y MANEJO:
No existe un tratamiento estandarizado para EMAD; el tratamiento empleado incluye esteroides, Inmunoglobulina Intravenosa (IgIV)) o plasmaféresis; sin embargo la mayoría de datos reportados en la literatura provienen de estudios con fallas metodológicas, reportes de casos o estudios con poblaciones pequeñas por lo que resulta difícil la extrapolación de los resultados.
ESTEROIDES:
Todas las series publicadas de EMAD describen buena respuesta terapéutica a los corticoides en dosis altas. Sin embargo están descritos una gran variedad en el tipo, dosis, ruta de administración, y esquema de utilización. Existe un primer reporte de EMAD publicado en 1953 usando ACTH. Mas tarde en la época pre-RMN se utilizaron prednisona, corticotropina o dexametasona con mejoría tanto en niños como adultos. Varios pacientes de estos reportes tuvieron recurrencia de la sintomatología al descontinuar el esteroide mejorando al restituirlo (1,20,21,23).
Muchos grupos de investigadores pediátricos describieron el uso de altas dosis de esteroides usando la metilprednisolona en dosis de 10 a 30 mg/kg/día con un máximo de 1 gramo/día o dexametasona a razón de 1 mg/kg/día por 3 a 5 días seguido por esteroides orales de 4 a 6 semanas en esquema piramidal, con mejoría reportada en el 50 a 80% de los pacientes. Generalmente la mejoría se evidencia a las 24 a 48 horas del inicio del tratamiento esteroidea. En los casos más graves o de diagnostico tardío se acepta prolongar el tratamiento endovenoso hasta completar dos semanas. Existen varias comunicaciones de remisión espontánea en casos de EMAD (1,23).
INMUNOGLOBULINA INTRAVENOSA:
Hay múltiples casos reportados de uso de IgIV usada sola o combinada con esteroides. En algunos estudios, la IgIV fue administrada después de no obtener mejoría tras la administración de esteroides o en casos de recurrencia. La dosificación empleada es de 1 a 2 gr/kg, administrado en una dosis única o fraccionada en 3 a 5 días; sólo se observó respuesta en el 50% de los niños tratados. La efectividad de la IgIV se ha comunicado en un número reducido de publicaciones (1,60,61).
PLASMAFÉRESIS:
El uso de la plasmaféresis ha sido reportado en pequeño número de casos en los que fracasa el esteroide, sin embargo sólo excepcionalmente se ha mencionado en la literatura como tratamiento eficaz (1,62).
Actualmente la metilprednisolona es el tratamiento de elección, y la inmunoglobulina es la terapia de segunda línea cuando los corticoides fallan o están contraindicados.
EVOLUCIÓN Y PRONÓSTICO:
La EMAD generalmente tiene buen pronóstico en el niño. Esto es algo señalado en todas las series clínicas revisadas con un porcentaje bajo, aunque significativo, de secuelas (1,21). La EMAD generalmente es monofásica, pero existen pacientes con evolución recurrente o multifásica, por lo cual se deberían utilizar los criterios establecidos por el grupo de trabajo de EMP y trastornos relacionados para su adecuada ubicación. Característicamente las recidivas en la EMAD ocurren antes de los seis meses, sin embargo hay numerosas comunicaciones que describen recidivas en diferentes áreas del SNC y que ocurren uno o dos años e incluso varios años después del primer episodio (38).
Los factores que pudiesen incidir en mayor riesgo de recidivas estarían el no recibir tratamiento con corticoides, el no completar tratamiento con esteroides orales de cuatro a seis semanas después del tratamiento intravenoso. Un elemento importante para establecer el pronóstico es diferenciar EMAD de EMP. Existen estudios que describen que un 9% de una serie infantil de EMAD evolucionó a EMP; otros estudios señalan un porcentaje más alto, pero establecen las recidivas como confirmación para EMP (58).
Existen factores que reducen significativamente el riesgo de EMP después de EMAD, como serían: edad prepuberal, pródromo febril, alteración de la conciencia o letargia, trastornos del movimiento y convulsiones. Por otro lado, los factores que elevan el riesgo de EMP después de EMAD serían: edad post-puberal, ausencia de pródromos, mayor afectación sensorial, elevación del índice de IgG (se ha descrito 98% de riesgo para EMP) (58,59). Las secuelas se producen en alrededor del 20% de los casos, pero el porcentaje de discapacidad grave (cuatro o más según la escala de Kurzke) es bajo. En general las secuelas son motoras, pero hay estudios que describen alteraciones emocionales, conductuales y cognitivas en forma significativa (63-65).
CONCLUSIONES Y RECOMENDACIONES:
La EMAD es una enfermedad inflamatoria de naturaleza autoinmune, de curso la mayoría de las veces favorable. Los nuevos criterios propuestos por el grupo de trabajo de EMP y trastornos relacionados permitirán unificar los casos de pacientes con EMAD y poder caracterizar la evolución dentro de un concepto único para la adecuada interpretación y seguimiento. En la actualidad no hay marcadores biológicos, sin embargo la historia clínica y los hallazgos radiológicos a nivel de la RMN nos permiten tener un adecuada aproximación al diagnóstico de EMAD. Tenemos un alto grado de certeza de que el tratamiento de primera línea son los esteroides en altas dosis y dentro de un esquema de dosificación adecuado y completo para el niño. Deberá realizarse el esfuerzo para el seguimiento de estos niños, tanto clínico como a nivel de neuroimagen, lo que permitirá establecer definitivamente el diagnóstico de EMAD y descartar otros compromisos o entidades como la EMP. Es importante estimular a la población médica latinoamericana para establecer si la presentación epidemiológica, clínica, radiológica y respuesta al tratamiento se corresponde con otras latitudes.
REFERENCIAS:
1. Tenembaum S, Tanuja C, Ness J, Hahn J. Acute disseminated encephalomyelitis. Neurology 2007; 68(Suppl. 2):S23-S36. [ Links ]
2. Dale R. Acute Disseminated Encephalomyelitis. Pediatr Infect Dis 2003; 14(2):90-95. [ Links ]
3. Wingerchuk DM. Postinfectious encephalomyelitis. Curr Neurol Neurosci Rep 2003;3(3):256-264. [ Links ]
4. Silvia MT, Licht DJ. Pediatric central nervous system infections and inflammatory white matter disease. Pediatr Clin North Am 2005; 52:1107-1176. [ Links ]
5. Murthy SN, Faden HS, Cohen ME, et al. Acute disseminated encephalomyelitis in children. Pediatrics 2002; 110(2pt 1): e21. [ Links ]
6. Stüve O, Zamvil SS. Pathogenesis, diagnosis, and treatment of acute disseminated encephalomyelitis. Curr Opin Neurol 1999; 12:395-401. [ Links ]
7. Stonehouse M, Gupte G, Wassmer E, Whitehouse WP. Acute disseminated encephalomyelitis: recognition in the hands of general pediatricians.Arch Dis Child 2003; 88:122-4. [ Links ]
8. Leake JA, Albani S, Kao AS, Senac MO, Billman GF, Nespeca MP, et al. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J 2004; 23:756-764. [ Links ]
9. Rust R. Multiple sclerosis, acute disseminated encephalomyelitis, and related conditions. Semin Pediatr Neurol 2000;7: 66-90. [ Links ]
10. Legido A, Tenembaum S, Katsetos CD, Menkes JH. Autoimmune and postinfectious diseases. In: J.H. Menkes, H.B.Sarnat (eds). Child neurology. Lippincott Williams & Wilkins. Philadelphia 2005, pp. 557-657. [ Links ]
11. Dale RC, Branson JA. Acute disseminated encephalomyelitis or multiple sclerosis: can the initial presentation help in establishing a correct diagnosis? Arch Dis Child 2005; 90: 636-639. [ Links ]
12. Rust RS, Menkes JH. Autoimmune and postinfectious diseases. In: J.H. Menkes, H.B.Sarnat (eds). Child neurology. Lippincott Williams & Wilkins. Philadelphia 2000, pp. 627-691. [ Links ]
13. Bennetto L, Scolding N. Inflammatory post-infectious encephalomyelitis. J Neurol Neurosurg Psychiatr 2004; 75 (Suppl. 1):22-28. [ Links ]
14. Davis LE, Boss J. Acute disseminated encephalomyelitis in children: a changing picture. Pediatr Infect Dis J 2003; 9:829-831. [ Links ]
15. Glabinski AR, Bielecki B, Ransohoff RM. Chemokine upregulation follows cytokine expression in chronic relapsing experimental autoimmune encephalomyelitis. Scand J Immunol 2003; 58:81-88. [ Links ]
16. Allen IV. Demyelinating diseases. In: J.H. Adams, J.A.N. Corsellis, L.W. Duchen (eds). Greenfields neuropathology. 4 ed. Edward Arnold. London1984, pp. 338-384. [ Links ]
17. Lipton HL. Theilers virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun 1975; 11:1147-1155. [ Links ]
18. Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Ann Rev Immunol 1990; 8:579-627. [ Links ]
19. Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodríguez M. CD4+ and CD8+ T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol 1998; 72:7320-7329. [ Links ]
20. Erazo-Torricelli R. Encefalomielitis aguda diseminada en la niñez. Rev Neurol 2006; 42(Suppl. 3):S75-S82. [ Links ]
21. Tenembaum S, Chamoles N, Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology 2002;59:1224-1231. [ Links ]
22. Krupp L, Banwell B, Tenembaum S. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007; 68(Suppl 2):S7-S12. [ Links ]
23. Peña, JA, Montiel-Nava C, Hernández F, Medrano E, Valbuena O, Cardozo J. Encefalomielitis aguda diseminada en niños. Rev Neurol 2002; 34:163-168. [ Links ]
24. Ito T, Watanabe A, Akabane J. Acute disseminated encephalomyelitis developed after acute herpetic gingivoestomatitis. Tohoku J Exp Med 2000; 192:151-155. [ Links ]
25. Revel-Vilk S, Hurvitz H, Klar A, Virozov Y, Korn-Lubetzki I. Recurrent acute disseminated encephalomyelitis associated with acute cytomegalovirus and Epstein-Barr virus infection. J Child Neurol 2000; 15: 421-424. [ Links ]
26. Mariotti P, Batocchi AP, Colosimo C, Catani P, Stefanini MC, Colitto F, et al. Positive PCR for enterovirus in the cerebrospinal fluid of a child with acute disseminated encephalomyelitis. J Neurol 2004; 251:1267-1269. [ Links ]
27. Sacconi S, Salviati L, Merelli E. Acute disseminated encephalomyelitis associated with hepatitis C virus infection. Arch Neurol 2001; 58:1679-1681. [ Links ]
28. Saitoh A, Sawyer MH, Leake JAD. Acute disseminated encephalomyelitis associated with enteroviral infection. Pediatr Infect Dis 2004; 23:1174-1175. [ Links ]
29. Sonmez FM, Ödemis E, Ahmetoglu A, Ayvaz A. Brainstem encephalitis and acute disseminated encephalomyelitis following mumps. Pediatr Neurol 2004;30:132-134. [ Links ]
30. Tan HT, Kilikaslan M, Ömbas Ö, Büyükavci M. Acute Disseminated encephalomyelitis following hepatitis A virus infection. Pediatr Neurol 2004; 30:207-209. [ Links ]
31. McKendrick MW, Sharack B. Acute disseminated encephalomyelitis temporally associated with Campylobacter gastroenteritis. J Neurol Neurosurg Psychiatr 2004; 75:788-793. [ Links ]
32. Dale RC, Church AJ, Cardoso F, Goddard E, Cox TC, Chong WK, et al. Post-streptococcal acute disseminated encephalomyelitis with basal ganglia involvement and autoreactive antibasal ganglia antibodies. Ann Neurol 2001; 50:588-595. [ Links ]
33. Ozawa H, Noma S, Yoshida Y, Sekine H, Hashimoto T. Acute disseminated encephalomyelitis associated with poliomyelitis vaccine. Pediatr Neurol 2000; 23:177-179. [ Links ]
34. Yong VW. Immunopathogenesis of multiple sclerosis. Continuum 2004; 10(6):11-27. [ Links ]
35. Laouini D, Kennou MF, Khoufi S, Dellagi K. Antibodies to human myelin proteins and gangliosides in patients with acute neuroparalytic accidents induced by brain-derived rabies vaccine. J Neuroimmunol 1998; 91:63-72. [ Links ]
36. Van Bogaert L. Post-infectious encephalomyelitis and multiple sclerosis: the significance of perivenous encephalomyelitis. J Neuropathol Exp Neurol 1950; 9:219-249. [ Links ]
37. Ramaswamy V, Sinclair DB, Wheatley BM, Richer L, Snyder T. Epilepsia partialis continua: acute disseminated encephalomyelitis or Rasmussens encephalitis? Pediatr Neurol 2005; 32:341-345. [ Links ]
38. Mariotti P, Batochi AP, Colosimo C, Lo Monaco M, Caggiula M, Collito F, et al. Multiphasic demyelinating disease involving central and peripheral nervous system in a child. Neurology 2003; 60:348-349. [ Links ]
39. Tenembaun Silvia. Disseminated encephalomyelitis. Clin Neurol Neurosurg. Article in Press. Elsevier 2008. [ Links ]
40. Baum PA, Barkovich AJ, Koch TK, Berg BO. Deep gray mater involvement in children with acute disseminated encephalomyelitis. AJNR 1994; 15:1275-1283. [ Links ]
41. Hynson JL, Kornberg AJ, Coleman LT, Shield L, Harvey AS, Kean MJ. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology 2001;56:1308-1312. [ Links ]
42. Gabis LV, Panasci DJ, Andriola MR, Huang W. Acute disseminated encephalomyelitis: an MRI/MRS longitudinal study. Pediatr Neurol 2004; 30:324-329. [ Links ]
43. Caldemeyer KS, Smith RR, Harris TM, Edward MK. MRI in acute disseminated encephalomyelitis. Neuroradiol 1994;36:216-220. [ Links ]
44. Kimura S, Nezu A, Ohtsuki N, Kobayashi T, Osaka H, Uehara S. Serial magnetic resonance imaging in children with postinfectious encephalitis. Brain Dev 1996; 18:461-465. [ Links ]
45. Khurana DS, Melvin JJ, Kothare SV, Valencia I, Hardison SS, Yum S, et al. Acute disseminated encephalomyelitis in children: discordant neurologic and neuroimaging abnormalities and response to plasmapheresis. Pediatrics 2005; 116:431-436. [ Links ]
46. Gener B, Garaizar-Axpe C, Ruiz-Espinoza C, Prats-Viñas JM. ¿Puede la encefalomielitis aguda diseminada cursar de forma diferida? Rev Neurol 2001; 32:1132-1135. [ Links ]
47. Idrissova ZR, Boldyreva MN, Dekonenko EP, Malishev NA, Leontyeva IN, Martinenko IN, et al. Acute disseminated encephalomyelitis in children: clinical features and HLA-DR linkage. Eur J Neurol 2003; 10:537-546. [ Links ]
48. Hung KL, Liao HT, Tsai ML. Postinfectious encephalomyelitis: etiologic and diagnostic trends. J Child Neurol 2000; 15:666-670. [ Links ]
49. Dale RC, de Sousa C, Chong WK, Cox TCS, Harding B, Neville BGR. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis. Brain 2000; 12:2407-2422. [ Links ]
50. Triulzi F, Scotti G. Differential diagnosis of multiple sclerosis: contribution of magnetic resonance techniques. J Neurol Neurosurg Psychiatr 1998;64 (Suppl):S6-S14. [ Links ]
51. Mikaeloff Y, Suissa S, Vallée L, Lubetzki C, Ponsot G, Confavreux C, et al. First episode of acute inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr 2004; 144:246-252. [ Links ]
52. Alper G, Schor NF. Toward the definition of acute disseminated encephalomyelitis of childhood. Curr Opin Pediatr 2004; 16:637-640. [ Links ]
53. Gómez-Hernández A, Herranz-Fernández JL, Arteaga-Maiyón-Cabeza RM, Holanda-Peña MS. Encefalomielitis aguda diseminada de evolución bifásica. Bol Pediatr 2003; 43:64-69. [ Links ]
54. Bassuk AG, Keating GF, Stumpf DA, Burrowes DM, Stack C. Systemic lymphoma mimicking acute disseminated encephalomyelitis. Pediatr Neurol 2004; 30:129-131. [ Links ]
55. Poser CM, Paty DW, Scheinberg L, Mc Donald WI, Compston A, Edan G, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocol. Ann Neurol 1983; 13:227-231. [ Links ]
56. McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001; 50:121-127. [ Links ]
57. Paty DW, Oger JJ, Kastrukoff LF, Hashimoto SA, Hooge JP, Eisen AA, et al. MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal bandings and CT. Neurology 1988;38:180-185. [ Links ]
58. Peña JA, Montiel-Nava C, Ravelo ME, González S, Mora-La Cruz E. Esclerosis múltiple en niños. Problemas específicos de diagnóstico. XIII Congreso y XVII Curso de la Academia Iberoamericana de Neurología Pediátrica (AINP). Filadelfia, 31 de marzo-2 de abril 2005. [ Links ]
59. Hahn J, Pohl D, Rensel M, Rao S. Differential diagnosis and evaluation in pediatric multiple sclerosis. Neurology 2007; 68(Suppl. 2):S13-S22. [ Links ]
60. Shahar E, Andraus J, Savitzki D, Pilar G, Zelnik N. Outcome of severe encephalomyelitis in children: effect of high-dose methylprednisolone and immunoglobulins. J Child Neurol 2002; 17:810-814. [ Links ]
61. Andersen JB, Rasmussen LH, Herning M, Paerregaard A. Dramatic improvement of severe acute disseminated encephalomyelitis after treatment with intra-venous immunoglobulin in a three year old boy. Dev Med Child Neurol 2001; 43:136-138. [ Links ]
62. Miyazawa R, Hikima A, Takano Y, Arakawa H, Tomomasa T, Morikawa A. Plasmapheresis in fulminant acute disseminated encephalomyelitis. Brain Dev 2001; 23:424-426. [ Links ]
63. Anlar B, Basaran C, Kose G, Guven A, Haspolat S, Yakut A, et al. Acute disseminated encephalomyelitis in children: outcome and prognosis. Neuropediatrics 2003; 34:194-199. [ Links ]
64. Kutzke JF. Rating neurologic impairment in multiple sclerosis: an Expanded Disability Status Scale (EDSS). Neurology 1983; 33:1444-1452. [ Links ]
65. Jacobs RK, Anderson VA, Neale JL, Shield LK, Kornberg AJ. Neuropsychological outcome after acute disseminated encephalomyelitis: impact of age at illness onset. Pediatr Neurol 2004; 31:191-197. [ Links ]

