










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Puericultura y Pediatría
versión impresa ISSN 0004-0649
Arch Venez Puer Ped v.73 n.2 Caracas jun. 2010
Leucemia linfoblástica aguda: Evaluación clínico terapéutica del protocolo total XV modificado. Hospital universitario de Caracas 2003-2007.
Zulay Nohemí Chona De Armas (*), Erika Francisca Montero Ávila (**), Joaquín José Inaty Lamillo (***)
(*) Pediatra – Hematóloga, Adjunto del Servicio de Hematología del Hospital Universitario de Caracas. Caracas, Venezuela
(**) Médico Internista - Hematóloga. Adjunto del Servicio de Hematología del Hospital Universitario de Caracas. Caracas, Venezuela
(***) Pediatra - Hematólogo, Adjunto del Servicio de Hematología del Hospital Universitario de Caracas. Caracas, Venezuela
Correspondencia: Dra. Zulay Nohemí Chona De Armas, Hospital Universitario de Caracas Servicio de Hematología 1er piso Ciudad Universitaria Los Chaguaramos Caracas, Venezuela. Telf. y Fax: 0212- 6067318 Celular: 0416-8177406. Correo: chonazulay@gmail.com
RESUMEN
Introducción: La leucemia linfoblástica aguda (LLA) se caracteriza por la proliferación clonal y acumulación de células linfoides malignas en médula ósea y en sangre periférica.
Objetivos: Identificar los aspectos clínico-hematológicos, evolución terapéutica y morbimortalidad en niños con LLA de novo tratados con el Protocolo Total XV modificado, en el Servicio de Hematología del Hospital Universitario de Caracas (HUC) entre 2003-2007.
Métodos: Estudio clínico-epidemiológico, descriptivo y retrospectivo mediante la revisión de historias clínicas de pacientes menores de 18 años.
Resultados: Los síntomas clínicos al diagnóstico fueron hipertermia, astenia, cefalea, hiporexia, sangrado y dolor óseo; los signos: adenopatías, hepatoesplenomegalia y fiebre; mayor prevalencia en el género masculino: 64,7% y entre 1 a 10 años (67,7%). La mayoría presentó anemia, leucocitosis y trombocitopenia. La infiltración del SNC fue del 5,9%. Se obtuvo un 79,4% de remisión completa (RC) en la fase de inducción, la morbilidad principal fue por neutropenia febril y 8,7% de mortalidad. En la fase de consolidación, se mantuvo la tasa de RC (79,9%), la morbilidad fue por hepatotoxicidad y 6,8% de mortalidad. En la fase de mantenimiento, se mantuvo la tasa de RC 80% pero se presentó un 11,6% de recaídas, mayor morbilidad infecciosa y 19,2% de mortalidad. La sobrevida global (SG) y la sobrevida libre de enfermedad (SLE) con una mediana de seguimiento de 24 meses, fue: 57% y 18,8%, respectivamente.
Conclusión: La estrategia para adaptar el Protocolo Total XV modificado en el Servicio de Hematología, no fue efectiva para mejorar la SG ni SLE al compararlo con la literatura internacional.
Palabras clave: leucemia linfoblástica aguda, sobrevida global y sobrevida libre de enfermedad.
SUMMARY
Introduction: Acute lymphoblastic leukemia (ALL) is characterized by clonal proliferation and accumulation of malignant lymphoid cells in bone marrow and peripheral blood.
Objectives: To identify clinical and hematological aspects, therapeutic outcome and morbid mortality in children with de novo ALL treated with the modified Total Protocol XV, in the Department of Hematology, Hospital Universitario de Caracas (HUC) between 2003- 2007.
Methods: Clinical and epidemiological, descriptive, retrospective study by reviewing medical records of patients under 18 years.
Results: Clinical symptoms at diagnosis were hyperthermia, fatigue, headache, anorexia, bleeding and bone pain. Signs were lymphadenopathy, hepatosplenomegaly and fever, more prevalent in male 64.7% and in patients between 1 and 10 years (67.7 %). Most had anemia, leukocytosis and thrombocytopenia. CNS infiltration was present in 5.9%. We obtained a 79.4% complete remission (CR) in the induction phase, the major morbidity was febrile neutropenia and 8.7% mortality. In the consolidation phase, CR rate remained the same (79.9%), morbidity was 6.8% for hepatotoxicity and mortality. In the maintenance phase, CR rate was 80% but there was an 11.6% relapse, and the infectious morbidity and mortality rate increased to 19.2%. Overall survival (OS) and disease-free survival (DFS) with a median follow-up of 24 months was 57% and 18.8% respectively.
Conclusion: The strategy to adapt the Total Protocol XV modified in the Hematology Department was not effective in improving the OS and SLE when compared with international literature.
Keywords: acute lymphoblastic leukemia, overall survival and disease-free survival.
Recibido 28/3/2010 Aceptado:10/6/2010:
INTRODUCCIÓN
Las leucemias agudas son un grupo heterogéneo de neoplasias que afectan la Stem Cell (SC) es decir, las células progenitoras hematopoyéticas no comprometidas o parcialmente comprometidas. La enfermedad fue descrita por
primera vez en 1827 por Velpeau y definida como leucemia por Virchow en 1845. De acuerdo a la célula de origen, las leucemias se clasifican en leucemias linfoblásticas y mieloblásticas (1).
La Leucemia Linfoblástica Aguda (LLA) se caracteriza por la proliferación clonal y acumulación de células linfoides malignas en la médula ósea (MO) y en la sangre periférica, es la más común en niños sobre todo entre los 2 y los 5 años, y la incidencia es ligeramente superior en el género masculino, con una relación 1,3 a 1 (2). Constituye el 25% de todos los cánceres en la edad pediátrica y aproximadamente 75% de todos los casos de leucemia en la infancia (3). La incidencia es de 3-4 casos por cien mil niños en EE.UU (2), similar a la encontrada en Colombia en los últimos años (4). En Venezuela no se dispone de datos publicados sobre la incidencia de la enfermedad; no obstante, el registro central de cáncer del Programa Nacional de Oncología del Ministerio del Poder Popular de la Salud (MPPS) ofrece información que resulta de ayuda. Para el quinquenio 1995-1999, la notificación de patologías oncológicas de acuerdo a la localización topográfica, ubica a las leucemias en general (agudas y crónicas) en el séptimo lugar, con una tasa de 3,31 por 100.000 habitantes. La prevalencia total calculada, es de 1,45 por 100.000 habitantes, con una relación LLA/LMA de 4:1 en los menores de 19 años. Según los datos estadísticos publicados en el Anuario Epidemiológico de 2005 (el más actualizado hasta la fecha) y los datos de morbilidad del Registro Central de Cáncer, del Programa de Oncología del MPPS, las leucemias son más frecuentes en la población masculina, con una incidencia estimada de 819 casos anuales y una mortalidad estimada de 460 muertes al año; en cuanto al género femenino, la incidencia estimada es de 721 casos al año y una mortalidad estimada de 405 muertes al año para el año 2007. Por otra parte, la incidencia anual de casos de cáncer en niños y jóvenes menores de 15 años es dominada por las leucemias (más de 600 casos anuales), representando el 40% para el año 2006 (5).
La etiología de la enfermedad no se conoce exactamente, pero se han implicado una serie de factores, entre los cuales las alteraciones citogenéticas adquiridas parecen jugar un papel muy importante. Estas alteraciones incluyen básicamente cambios cuantitativos en el número de cromosomas, aberración en la expresión de los proto-oncogenes y traslocaciones cromosómicas, que crean genes de fusión que codifican kinasas activas, con subsiguiente alteración en los factores de trascripción, lo que puede contribuir a la transformación leucémica de la SC hematopoyética o de los progenitores comprometidos por alteración en el proceso de regulación, produciendo una capacidad de autorrenovación ilimitada o en el control de la proliferación, bloqueando la diferenciación y promoviendo la resistencia a las señales de muerte celular (6).
En cuanto al tratamiento, la LLA es una neoplasia rápidamente fatal si no se trata. Actualmente, cuatro de cada cinco niños pueden ser curados con los protocolos de tratamiento utilizados. Éste se divide en tres fases, a saber: Inducción, Consolidación y Mantenimiento, durante las cuales se incorpora la profilaxis del sistema nervioso central (SNC), con quimioterapia intratecal (QT IT) o altas dosis de quimioterapia (7). La Inducción tiene como objetivo erradicar más del 99% de la masa inicial de células leucémicas, restaurar la hematopoyesis normal y la condición física del paciente. Esta fase del tratamiento incluye la administración de glucocorticoides, siendo la dexametasona la más utilizada por tener mejor penetración al SNC, asociada a vincristina, L-asparaginasa y un antracíclico. Con la inducción, usualmente más del 90% de los pacientes alcanzan remisión completa (RC) (8).
La fase de consolidación mejora la remisión alcanzada por la inducción; comúnmente los regímenes incluyen altas dosis de metotrexate asociado a 6–mercaptopurina. La fase de mantenimiento tiene como objetivo mantener la remisión ya alcanzada. La mayoría de los regímenes contempla la combinación de metotrexate semanal con 6–mercaptopurina diaria. La duración total del tratamiento debe ser de 2 años o de 2 años y seis meses (9,10). Las complicaciones asociadas al tratamiento pueden ser agudas: síndrome de lisis tumoral, mucositis, sangrado, infecciones bacterianas y fúngicas asociadas a mielosupresión prolongada son frecuentes, toxicidad cardíaca y hepática, trombosis, pneumonitis. Las complicaciones tardías de mayor significancia son las disfunciones orgánicas, infertilidad, segundas neoplasias, hepatitis crónica, problemas músculos-esqueléticos y alteraciones de la función cognitiva: cuando se utilizaba radioterapia (RT) profiláctica al SNC y también se observaba frecuentemente cataratas y disfunciones endocrinas (11-13).
Los grandes avances en el éxito del tratamiento de la LLA comprenden un período de 50 años, durante el cual ha dejado de ser una condición uniformemente fatal, para constituirse en una enfermedad con una tasa de curación aproximadamente del 85% en los países desarrollados (8). Los progresos realizados en la década de 1990 con relación a la caracterización molecular, cariotipo e inmunofenotipo de los blastos leucémicos han mejorado la comprensión de la biología de la LLA y ha refinado los criterios de clasificación de riesgo, lo cual permite la asignación adecuada de los pacientes a diferentes esquemas de tratamiento. La combinación de estos avances, sumada a la continua mejoría en las medidas de soporte, ha producido la tasa de curación mencionada (8). Sin embargo, en países en vías de desarrollo como Venezuela, las limitaciones económicas y las políticas de salud no han permitido implementar en todos los centros la infraestructura necesaria para brindar el manejo óptimo a estos pacientes.
Desde el inicio del tratamiento los progresos terapéuticos han sido sustanciales, y en ello ha jugado un papel preponderante los aportes realizados por diversos centros, especialmente por el St. Jude Children´s Research Hospital (Menphis, USA) con una serie de protocolos clínicos sucesivos iniciados en 1962, cada uno de ellos diseñados con la finalidad de mejorar los resultados obtenidos con el precedente. Uno de sus protocolos más recientes es el Total XV, con el cual han logrado índices de Sobrevida Libre de Enfermedad (SLE) del 85,6% y SG del 93,5% a los 5 años (14). Este protocolo Total XV del St. Jude Children´s Research Hospital fue modificado para su adaptación a países en vías de desarrollo como Venezuela, por el Grupo Cooperativo Caracas–Maracaibo para pacientes con LLA, dentro de las modificaciones destacan la administración de dosis menores de metotrexate y de citarabina y no se realiza la evaluación de enfermedad mínima residual (EMR) al final de la inducción.
El objetivo del presente trabajo es señalar los aspectos clínico-hematológicos, la evolución y la morbimortalidad de 34 niños con LLA de novo, tratados en el Servicio de Hematología del Hospital Universitario de Caracas (HUC) con el Protocolo de Quimioterapia Total XV modificado, usado durante el período 2003-2007 y compararlo con las obtenidas con protocolos previamente utilizados en el HUC y por grupos internacionales.
MÉTODOS
Se realizó un estudio clínico-epidemiológico descriptivo y retrospectivo desde septiembre de 2003 hasta septiembre de 2007, a través de la revisión de historias clínicas de pacientes menores de 18 años, con diagnóstico de LLA de novo y tratados con el Protocolo de Quimioterapia Total XV modificado en el Servicio de Hematología del Hospital Universitario de Caracas (HUC).
Se diagnosticaron 34 niños con LLA de novo con edades comprendidas entre 1 y 18 años, para la clasificación morfológica se siguieron los criterios del grupo Francés- Americano-Británico (FAB) (1); y recibieron tratamiento con el Protocolo de Quimioterapia Total XV modificado, el cual clasifica a los pacientes según el riesgo basándose en la edad, cifras de glóbulos blancos, la presencia o ausencia de enfermedad en el SNC, el índice de ADN y el cariotipo al momento del diagnóstico. En este estudio no se realizan los 2 últimos parámetros por no tener disponibilidad ni acceso a ellos.
1. Bajo Riesgo (BR):
-Edad: >1 año <10 años
-Glóbulos blancos (GB) < 50 x 109/L
-Ausencia de marcadores de células T
-Ausencia de masa mediastinal
-Ausencia de infiltración testicular
-SNC 1: ausencia de blastos en el LCR (cytospin)
-Índice de ADN > 1,16 o < de 1,60
-Ausencia de t (9;22) o t (1;19)
2. Alto Riesgo (AR):
-Edad: <1 año >10 años
-GB > 50 x 109/L
-Inmunofenotipo de células T
-Presencia de masa mediastinal
-Infiltración testicular
-SNC 2: < 5 GB/μL con blastos detectables en el LCR o SNC 3: > 5 GB/μL con blastos detectables en el LCR y/o parálisis de nervios craneales.
-Índice de ADN < 1,16 o > de 1,60
-Presencia de t (9;22) o t (1;19)
-Pacientes con persistencia de blastos (más del 5%) en el aspirado de MO realizado en el día 15 y/o el día 36.
El protocolo consta de tres fases: Inducción, Consolidación y Mantenimiento; las cuales se establecen de acuerdo al riesgo: AR o BR al momento del diagnóstico:
La fase de inducción tiene una duración de 36 días y contempla el uso de prednisona (40mg/m²/día), daunoblastina (25mg/m²/dosis), vincristina (1,5mg/m²/dosis), L-asparaginasa (10.000Ud/m²/dosis), ciclofosfamida (1gr/m²/dosis), citarabina (75mg / m² / dosis), 6–mercaptopurina (60mg/m²/dosis), QT IT triple profiláctica o terapéutica según la edad (ver Tabla 1), aspirado de MO el día 15 (en la cual si hay persistencia de blastos >5% el paciente se pasa a la categoria de AR, contemplando una dosis adicional de daunoblastina y tres adicionales de L-aspaginasa y se continua el protocolo de AR) y el día 36; siendo mayor el número de dosis aplicadas de L-asparaginasa y QT IT triple profiláctica o terapéutica en los pacientes de AR.

La fase de consolidación tiene una duración de 14 días y se caracteriza por el uso de altas dosis de metotrexate el día 1 y el día 8 (2gr/m²/dosis para BR y 3gr/m²/dosis para AR) asociado a 6–mercaptopurina (75mg/m²/día) por 14 días y QT IT triple profiláctica el día 1 y 8 para ambos grupos.
La fase de mantenimiento es de 120 semanas para las niñas y de 146 semanas para los niños, se caracteriza por el uso de 6–mercaptopurina diaria y metotrexate intramuscular (IM) semanal con pulsos de dexametasona y vincristina cada 4 semanas, una fase de reinducción en la semana 7 y altas dosis de metotrexate (2gr/m²/dosis) cada 8 semanas durante el primer año, e igualmente QT IT triple cada 8 semanas hasta la semana 56 durante el primer año y estudio de LCR sin QT IT cada 16 semanas durante el segundo año, con evaluación de MO en las semanas 10, 31, 56, 120 y 146 en los pacientes de BR. Los pacientes de AR reciben igualmente lo antes mencionado adicionándose ciclofosfamida, citarabina, una fase de reinducción más en la semana 17; altas dosis de metotrexate a 3gr/m²/dosis cada 8 semanas durante el primer año, e igualmente QT IT triple cada 4 semanas hasta la semana 68, y estudio de LCR sin QT IT cada 16 semanas durante el segundo año hasta la omisión del tratamiento. Si el paciente presenta SNC 2 al diagnóstico, igualmente seguirá el esquema de QT IT triple antes mencionado; pero si cursa con SNC 3 al diagnóstico, recibirá QT IT triple cada 4 semanas durante el primer año de mantenimiento, seguido en la semana 56 por RT craneal 1800 cGy dividida en 12 secciones, a razón de 150 cGy/diarios más una dosis de QT IT triple semanal por 5 semanas (57, 58, 59, 60, 61), después de la RT craneal no se realizará más tratamiento al SNC (en niños menores de 2 años, la RT se cumplirá después de los 2 años). El aspirado de MO se realiza en las semanas 10, 20, 31, 55, 120 y 146 en el caso de los varones.
Conductas ante algunos eventos:
1. Recaída hematológica: los pacientes con >25% de linfoblastos en el aspirado de MO, deben ser retirados del estudio e incluirlos en protocolos de recaídas.
2. Recaída en el SNC con remisión hematológica: se define como la existencia de >5 GB/μL con blastos en el LCR en exámenes por cytospin (sólo SNC 3). Estos pacientes permanecerán en el estudio:
a. Si la recaída es antes de la semana 56 de la terapia de mantenimiento, se administra un curso de reinducción y QT IT triple 1 semanal por 4 a 6 semanas, con un mínimo de dos adicionales luego que haya ocurrido el clearence de los blastos del LCR. Al alcanzarse la remisión del SNC, el paciente debe ser llevado a RT cráneo-espinal temprana (Craneal: 2400cGy y Espinal: 1500 cGy). En caso de niños menores de 2 años, debe retrasarse hasta que sea mayor de 2 años, mientras tanto debe cumplirse QT IT triple cada 4 a 8 semanas.
b. Si la recaída ocurre después de la semana 56 de la terapia de mantenimiento, se administra un curso de reinducción y QT IT triple 1 semanal por 4 a 6 semanas, con un mínimo de dos adicionales, luego que haya ocurrido el clearence de los blastos del LCR. Para pacientes que no han sido irradiados se cumple RT cráneo-espinal como (a) seguido de tratamiento de reinducción. El tratamiento de mantenimiento se reinicia por al menos un año. Para pacientes que han sido irradiados, el segundo ciclo de RT se pospone para seis meses después del primer ciclo.
3. Si los pacientes presentan un SNC 2, no serán considerados con recaída al SNC manifiesta, continuarán con QT IT mensual y no recibirán RT.
4. Los niños con cualquier otra forma de recaída extramedular serán retirados del estudio y son elegibles para cualquier otro protocolo de recaída.
La respuesta al tratamiento se evaluó por la obtención de Remisión Completa (RC), Sobrevida Libre de Enfermedad (SLE) y Sobrevida Global (SG). La RC se definió como la regresión de todos los síntomas y signos, la desaparición de toda evidencia hematológica de enfermedad con menos del 5% de células leucémicas y la restauración de la función normal de la MO con recuperación de las series eritrocítica, granulocítica (>1x109/L neutrófilos en sangre periférica) y megacariocítica (>100x109/L de plaquetas en sangre periférica) y Remisión Parcial (RP) se definió como la persistencia de blastos entre 5 y 15% en la MO y la progresión de la enfermedad por la persistencia o empeoramiento de la enfermedad, a pesar del tratamiento. La recaída se definió como la reaparición de blastos en sangre periférica con evidencia de más de 5% de blastos en la MO o extramedular (infiltración del SNC o testicular). La muerte temprana es aquélla que ocurrió por cualquier causa antes del tratamiento y durante las primeras seis semanas del mismo. La SLE se determinó partiendo desde el momento de alcanzar la RC hasta la aparición de cualquier evidencia de actividad leucémica medular o extramedular. La SG se estimó desde el diagnóstico hasta la muerte o la última observación.
Para el análisis estadístico, se realizó el cálculo de frecuencias y proporciones en cada una de las variables cualitativas u ordinales, y para las variables cuantitativas el cálculo de frecuencias, promedios, rangos y desviación estándar. Se aplicó la prueba de Chi cuadrado expresada en términos de probabilidades, con intervalo de confianza de 95% y un error del 5%; para determinar si hubo o no diferencia significativa entre las variables estudiadas. La estimación de la sobrevida para cada grupo se basó en el análisis actuarial de Kaplan – Meier, los contrastes de las funciones de supervivencia se realizaron usando la prueba long rank (15), según la distribución de Chi cuadrado. Se consideró como valor significativo p < 0,05 y altamente significativo p < 0,01.
RESULTADOS
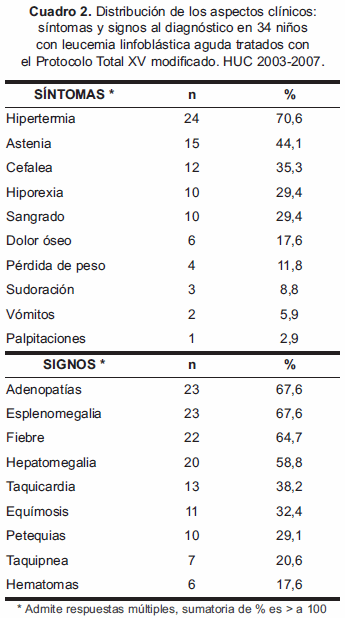
El diagnóstico de LLA se estableció en base a los parámetros clínicos y hematológicos aceptados internacionalmente. De los 34 niños, 64,7% pertenecían al género masculino y 35,3% al femenino, con una relación M/F: 1.8:1; el 67,7% tenían edades comprendidas entre 1 y 10 años. Los síntomas referidos al momento del diagnóstico, así como los hallazgos al examen físico fueron diversos y se señalan por orden de frecuencia en el Cuadro 2.
Los aspectos hematológicos para el momento del diagnóstico revelaron un valor promedio de hemoglobina de 8,8 gr/dl 2,5 (3,2 a 14 gr/dl), hematocrito 26,6% 7,5 (9,4 a 43,1%), glóbulos blancos 25 x 109/L 36,3 (1,6 a 175 x 109/L) y plaquetas 119,5 x 109 /L 111,3 (6 a 423 x 109 /L).
A todos los pacientes se les realizaron pruebas de función hepática y renal, serologías, pruebas de coagulación, ecocardiograma, aspirado y biopsia de MO, estudio del líquido céfalo raquídeo (LCR). El inmunofenotipo de MO se realizó en el 100% de los pacientes y fue compatible con el diagnóstico de LLA Común (TDT +, HLA-DR +, CD19 +, CD24 -, CD10 +, CD20 -, rearreglo de genes Ig +, Ig citoplasmática y de membrana negativas), la citoquímica PAS+ se encontró reportada sólo en el 2,9% de los casos y el estudio de biología molecular (oncogenes) en el 14,7% de los casos. La clasificación morfológica se realizó según los criterios del FAB, fue descrita en el 14,7% de los casos como blastos tipo L2, en el 5,9% de los casos blastos tipo L1 y en el resto no se especificó la variedad morfológica. El estudio del LCR al momento del diagnóstico se realizó en el 100% de los casos, evidenciándose infiltración de SNC 2 en 2 niños (5,9%) (Cuadro 3).

Al evaluar de la respuesta terapéutica en las diferentes fases del protocolo se observó: En la fase de inducción al realizarse el aspirado de MO correspondiente al día 15, se encontró RC en 4 niños (11,8%) de BR y en 11 niños (32,3%) de AR, persistencia de blastos en 13 niños (38,2%) de BR (quienes se reclasificaron y se les asignó como AR por persistencia de blastos en el día 15 según el protocolo) y en 4 niños (11,8%) de AR, no fue posible realizarla en 2 niños de AR por muerte temprana. En la evaluación de MO del día 36, la cual correspondía a la culminación de la fase de inducción, 27 niños (79,4%) alcanzaron RC, de los cuales 4 niños (11,8%) eran de BR y 23 niños (67,6%) eran de AR, 2 niños de AR alcanzaron RP (5,9%), 2 niños de AR no obtuvieron respuesta al tratamiento (5,9%) siendo excluidos del protocolo y 3 niños (8,8%) fallecieron. Durante esta fase, la morbilidad estuvo representada por: neutropenia febril 11,7%, neuropatía por vincristina y hepatotoxicidad 5,8% cada una; mucositis, sepsis, ACV isquémico, hipofibrinogenemia, herpes zóster y neumocitosis 2,9% cada una.
De los 34 pacientes, 29 iniciaron la fase de consolidación, la cual comprendió las altas dosis de metotrexate en el día 8 y 14, evidenciándose en la bioquímica hepática del día 8 sólo elevación de aminotransferasas; y el día 14 elevación de aminotreanferasas en mayor grado e hiperbilirrubinemia. Durante esta fase, se observó seroconversión para virus de hepatitis C (VHC) en un niño (3,4%). Al evaluar la respuesta del tratamiento en esta fase se observó RC continuada en 26 niños (89,7%): 4 niños (13,8%) de BR y 22 niños (75,9%) de AR; 2 niños (6,9%) fallecieron: uno por síndrome de piel escaldada y otro por bacteriemia por A. Woffi; 1 niño (3,4%) fue excluido del protocolo por toxicidad neurológica. Se observó hepatotoxicidad en el 27,6% de los niños y neutropenia febril en el 3,4% de los niños. Iniciaron fase de mantenimiento 26 niños, observándose, en algún momento de esta fase, morbilidades como: neutropenia febril en 96,5%, infección del tracto respiratorio en 37,9%, mucositis, constipación por vincristina, infección de partes blandas, hepatotoxicidad e insuficiencia hepática en 6,9% cada una de ellas, fisura anal y síndrome diarreico agudo en 3,4% cada uno. Se evidenció seroconversión para el VHC en 5 niños (17,2%). La evaluación de esta fase hasta el mes de septiembre de 2007 evidenció recaída en 5 niños (19,2%) distribuidos así: 3 niños con SNC 3 (11,6%), 1 niño en MO y SNC (3,8%), 1 niño en MO (3,8%); el tiempo de la presentación de la recaída fue: uno antes de los 12 meses (3,8%), uno antes de los 24 meses (3,8%) y 3 después de los 24 meses (11,6%) en un período de seguimiento de 50 meses; los 2 con recaída medular fueron incluidos en otro protocolo y los 3 niños con recaída en el SNC recibieron RT cráneo espinal, de acuerdo a lo establecido en el protocolo y alcanzaron RC (Cuadro 4). Las causas de mortalidad en cada una de las fases del protocolo se señalan en el Cuadro 5.


En total, se les hizo seguimiento a 34 niños desde septiembre 2003 hasta el 30 de septiembre de 2007 (50 meses de observación), el análisis actuarial señaló que la SG es de 73,1% y la SLE es de 31,3% a los 12 meses de seguimiento, la SG baja a 57% y la SLE baja a 18,8% a los 24 meses de seguimiento y disminuyó la SG a 19,2% y la SLE a 10,3% a los 50 meses de observación. El análisis estadístico indicó una diferencia significativa (p < 0,025); continuaron vivos 19 niños (56%) en RC hasta septiembre 2007 de los cuales 4 ya culminaron la fase de mantenimiento (Figura 1).

En cuanto a la SLE, según el grupo de riesgo se evidenció que fue de 75,3% para el grupo de BR y 67% para el grupo de AR a los 12 meses de seguimiento, la SLE fue de 65,2% para el grupo de BR y 54,2% para el grupo de AR a los 24 meses de seguimiento, y la duración promedio de la SLE para el grupo de BR fue de 51,1% y para el grupo de AR de 41,5% a los 50 meses de observación; el análisis estadístico indicó que hubo diferencia significativa (p < 0,035) (Figura 2).

DISCUSIÓN
El desarrollo de drogas antileucémicas de mayor efectividad, el mejor conocimiento de la biología de la enfermedad y la inclusión de una metodología científica cuantitativa para evaluar la respuesta a los agentes quimioterápicos, han permitido que en la actualidad, más del 90% de los niños con LLA se mantengan en RC prolongada e ininterrumpida por 5 años o más después del diagnóstico, y muchos se consideran curados (8). Tomando en cuenta los parámetros clínicos y la respuesta terapéutica, se encontró que mas de las dos terceras partes de los niños tenían edades comprendidas entre 1 y 10 años, esto coincide con lo reportado por el protocolo Total XV del St. Jude Children´s Research Hospital, con una media de 5,3 años (14) y por el Dana Faber Cancer Institute (DFCI) protocolo 95-01 (16). La literatura reporta que en el grupo de edades entre 2 y 6 años la sobrevida es 1,5 veces mayor que en los niños menores de 2 años o mayores de 10 años. El género masculino fue predominante en el presente estudio, al igual que lo señalado en la literatura, y esto tiene poco impacto en la inducción de la remisión y en la frecuencia de recaídas durante el primer año de tratamiento (17-20).
Entre los aspectos clínicos, los síntomas observados al diagnóstico coinciden con los descritos en la literatura. Los signos al diagnóstico coinciden con el estudio realizado en el HUC 1980-1985 (17) y con los estudios mexicano y chileno (20,21).
En relación a los parámetros de laboratorio al diagnóstico, en el hemograma se demostró afectación de las tres series, con una media de GB de 119,5 x 109/L, siendo esto considerado un factor de riesgo individual para determinar el grupo de riesgo al diagnóstico, posibilidad de remisión con la inducción, duración de la remisión y sobrevida de la enfermedad (14,19,20). En el estudio del LCR al diagnóstico, se evidenció infiltración del SNC en el 5,9% que correspondían a SNC 2, siendo esto reportado en la literatura en el 5% de los casos al diagnóstico, asociándose a baja tasa de remisión en inducción, alta tasa de recaída y corta sobrevida (22,23), lo cual fue evidenciado en este estudio.
En la revisión de 34 niños tratados con el Protocolo Total XV modificado en el Servicio de Hematología del HUC, en la mitad de los niños estudiados se evidenció la persistencia de blastos en el estudio de MO del día 15 de la inducción, de los cuales 13 niños eran de BR al diagnóstico y se reclasificaron como AR. En la MO realizada el día 36 de la inducción, se evidenció tasas de RC menores, RP y persistencia de enfermedad similares a los reportados por el Total XV del St. Jude Childrens Hospital, quienes evaluaron a 498 niños con edades entre 1 y 18 años, 492 niños obtuvieron RC (98,8%) con la inducción, 2 niños presentaron fracaso de inducción debido a infecciones fatales, 4 casos de leucemias refractarias y 3 de ellos fueron llevados a transplante alogénico de MO (24). El régimen de inducción con vincristina, prednisona/dexametasona, asparaginasa con QT intratecal, ha dado resultados en las tasas de RC de más del 95% (25). En pacientes de AR, un régimen de inducción más intenso con cuatro o cinco fármacos da un mejor resultado en la SLE (14, 26). En un estudio mexicano que trató a un grupo de 33 adolescentes con el protocolo pediátrico St Jude´s Total XI, la remisión inicial fue de 76% y la mediana de SLE fue de 29 meses (16). Más del 95% de los niños con LLA recién diagnosticada alcanzan RC en las cuatro primeras semanas de tratamiento. El 2 al 4% de los pacientes que no la alcanzan, 50% presentan muerte tóxica durante la inducción y el resto presentarán enfermedad persistente (16).
Las causas de muerte en esta fase son similares a las descritas en el estudio de 209 niños con LLA: experiencia de un grupo multicéntrico en Venezuela, y en el presente estudio la tasa de RC es menor a la reportada en la literatura, que se podría explicar por el tamaño de la muestra y mayor mortalidad en la fase de inducción (27).
En la fase de consolidación, que contempla alta dosis de metotrexate, el efecto adverso más importante fue la hepatotoxidad, mayor que la reportada en la experiencia del grupo multicéntrico en Venezuela 2% (27). Igualmente en esta fase, los resultados del presente estudio mostraron mayor frecuencia de recaídas y fallecimientos que lo reportado por el estudio del Total XV del St. Jude Childrens Hospital, ya que ellos sólo reportaron 3 recaídas post remisión y sólo un niño fallecido, que representa una tasa de mortalidad de 1,4%; sus 30 pacientes de BR permanecen libres de recaídas (24). Así mismo, es importante destacar que el Protocolo Total XV St. Jude Childrens Hospital demostró que con la intensificación de la QT sistémica y la IT triple, se presentaron tasas de infección diseminada por hongos y trombosis relativamente altas, y los niños mayores de 10 años de edad eran más propensos que los más jóvenes a desarrollar infección grave, osteonecrosis, trombosis e hiperglicemia, un hallazgo que podría explicarse por la disminución del aclaramiento de la dexametasona en los pacientes mayores. Cabe destacar, que ningún paciente murió de estas complicaciones gracias al cuidado, al apoyo precoz con antibióticoterapia de amplio espectro y la vigilancia estricta que caracteriza al St. Jude Childrens Hospital (24). Así mismo, demostraron que la irradiación craneal profiláctica puede ser totalmente omitida sin comprometer la supervivencia global. Con estas medidas, la tasa de recaída del SNC aislada fue de 2,7%, muy dentro del 1,5% al 4,5% del rango en los ensayos clínicos que utilizan irradiación craneal profiláctica como los estudios de Total XI, XII, XIIIA, XIIIB y XIV (14, 28, 29). Al comparar estos hallazgos con el protocolo Total XV modificado, en éste se presentó mayor variedad de comorbilidades infecciosas, que trajo como consecuencia una mayor tasa de mortalidad, e igualmente se obtuvo mayor tasa de recaída aislada en SNC.
Al comparar los resultados del Total XV St. Jude Childrens Hospital, la SLE a los 5 años y las estimaciones de SG fueron 85,6% (79,9% a 91,3%) y 93,5% (89,8% a 97,2%) respectivamente para todos los 498 pacientes, muy superiores a las de esta investigación, y como es de esperarse en los países desarrollados. El Protocolo Total XV St. Jude Childrens Hospital alcanzó una tasa de SG a los 5 años del 93,5%, que es superior a los resultados de todos los estudios importantes realizados hasta ahora en LLA en niños (14).
Se realizó un estudio en niños con LLA tratados con el protocolo BFM 86 en el Hospital J M de los Ríos entre 1998- 2001, donde se evaluó la eficacia y las complicaciones de dicho protocolo. Se incluyeron 50 niños, 31 fueron incluidos en el protocolo de AR y 20 fueron incluidos en el de BR. Todos alcanzaron RC posterior a la inducción, no se reportaron recaídas en SNC, la mayor complicación fue neutropenia severa febril. La primera causa de muerte fue sepsis y la segunda fue hemorragia. La SLE resultó en 68% para el grupo de BR y 70% para el grupo de AR (30), éstas superaron a las encontradas en este estudio.
Los resultados del Protocolo 2001 del Grupo Cooperativo Nacional (GCN), el cual se realizó entre el año 2000 y 2007, con un grupo de 224 niños con LLA con edades comprendidas entre los 13 meses y 18 años, se clasificaron en AR y riesgo estándar (RE). El tratamiento consistió en inducción, intensificación y mantenimiento. De los 224 niños: 108 niños eran RE (48%) y 116 niños eran de AR (52%), 11 niños murieron en la inducción (5%), 5 no alcanzaron la RC (2%), 208 niños alcanzaron RC (93%); en relación a las recaídas: 29 niños recayeron en MO (15%), 7 en SNC (4%), 2 en testículo (1%); 4 niños mueren en RC (2%) y 140 están vivos en RC (71%). La toxicidad más importante estuvo relacionada con la L-asparaginasa con 19 efectos adversos: 13 alergias (7%), 5 pancreatitis (2%) y 1 hiperglicemia. En comparación con los resultados de este estudio, se evidenció una mayor tasa de mortalidad en inducción y una menor tasa de RC en la fase de inducción; con respecto a las recaídas, en el presente estudio hubo una mayor prevalencia de infiltración al SNC, a pesar de cumplir QT IT desde el inicio de la fase de inducción, diferente a lo observado en este estudio, quienes inician QT IT profiláctica luego de alcanzar la RC postinducción. La SLE a los 5 años, con una mediana de seguimiento de 2 años, fue de 63% en RE y 65% en AR, parecida a los resultados de la presente investigación, a pesar de menor número de casos; la supervivencia libre de leucemia fue de 73% en RE y 73% en AR, y la SG fue de 87% en RE y 74% en AR (31), superiores a los hallazgos en la presente investigación. Se realizó un estudio retrospectivo en 102 pacientes con LLA pre B de novo en el Hospital de Especialidades Pediátricas de Maracaibo en el período 2003- 2007, donde evaluaron el protocolo Total XV modificado del St Jude que utilizan como Grupo Cooperativo; sus objetivos fueron describir las características observadas: edad, género, muertes y determinar la SG y SLE. Sus resultados fueron 60 niños (58,8%) del género masculino y 42 (41,2%) del género femenino, la edad promedio fue de 7,7 años y las muertes 18 (17,6%) y los vivos 84 (82,4%). Pacientes vivos en RC: 82 (97,6%) y pacientes vivos en 2da remisión: 2 (2,4%). Este estudio reveló una SG de 97,6% y una SLE de 82.4% (32), lo cual es superior en forma notable a las del presente estudio.
Comparando con grupos internacionales, los resultados son muy inferiores en cuanto a SG y SLE, probablemente debido a las facilidades que tienen los pacientes leucémicos en esos centros en cuanto a obtención de la quimioterapia, antibiótico- terapia y derivados sanguíneos (33).
Entre los estudios realizados en América Latina, el grupo colombiano encuentra SLE en el grupo de BR de 49% y en AR de 50%, long rank = 0,049 (19). En el grupo chileno la frecuencia de SLE global a los 5 años fue BR 75%, AR 62% y muy alto riesgo (MAR) 28%, con una mediana de seguimiento de 6,5 años. La incidencia acumulada de recaída en SNC fue 5,4% (34), lo cual coincide con lo encontrado en el presente estudio.
Al comparar con la literatura internacional, los resultados del ALL-BFM 90, con SLE de 78% con una media de observación de 4,8 años. La SLE fue de 85% en RE, 82% en RI. La tasa de recaída en SNC fue 0,8% y 1,5% para RI y AR, respectivamente. La tasa de mortalidad en inducción fue de 1%, principalmente por infección durante la neutropenia. Sin embargo, en Colombia, para mejorar el pronóstico de los niños con LLA tratados en el Instituto Nacional del Cáncer, usaron un protocolo basado en el BFM-90 y los regímenes convencionales (LSA2L2) que se implementaron desde 1993, con una cohorte de 123 pacientes se logró disminuir la tasa de muerte en inducción (7% vs 14%), pero un problema importante fue la tasa de RC aumento la de mortalidad del 5% al 14% (p < 0,06) (35), similar al estudio realizado; concluyen que una mejor terapia, atención, apoyo y mejoras en las condiciones socioeconómicas deben reducir la tasa de mortalidad de los países subdesarrollados en el futuro. Comparando esta tasa de mortalidad con otros centros como el Dana Faber Institute 85-01, fue 0,4% y 2,5% en el UKALL-X, lo cual no es comparable con la casuística del presente estudio, con una tasa de mortalidad de 8,7%. La tasa de mortalidad después de alcanzar RC fue de 1,6% en el ALL-BFM 90, de 3,4% en el UKALL-X y 3,6% en DFCI (26), comparando con el presente estudio en fase de consolidación fue 6,8% y 19,2% en mantenimiento. En el DFCI en niños entre 1 a 18 años, con una mediana de seguimiento de 6,5 años, la SLE fue 85% en el grupo de 1 a 10 años, 77% en el de 10 a 15 años y 78% en los de 15 a 18 años (36).
En conclusión, la estrategia para adaptar el Protocolo Total XV modificado no fue efectiva para mejorar la SG ni la SLE, al compararlo con la literatura internacional. La evaluación inicial de los pacientes con LLA debe realizarse con técnicas de laboratorio que generen información apropiada desde el punto de vista citogenético, inmunológico y molecular; y para su manejo óptimo requieren de un grupo multidisciplinario (Pediatra, Hematólogo, Nutricionista,
Psicólogo, Infectólogo, Odontólogo entre otros), de protocolos actualizados, de agentes citotóxicos efectivos y adecuadas medidas de soporte, incluyendo terapia transfusional, hallazgo y tratamiento precoz de las complicaciones infecciosas, atención cuidadosa a las necesidades metabólicas y nutricionales del paciente y soporte psicosocial permanente para el niño y su familia. Todo esto contribuye a la disminución de la tasa de mortalidad y a elevar la supervivencia a largo plazo.
RECOMENDACIONES
1.- Realizar de manera rutinaria a los pacientes con LLA, los estudios de biología molecular y estudios citogenéticos para conocer los oncogenes y/o las alteraciones cromosómicas, que permitan una adecuada estratificación del riesgo y aplicar las diferentes estrategias terapéuticas.
2.- Implementación de medidas de soporte más adecuadas, que incluyan: terapia transfusional óptima, antibióticoterapia de amplio espectro, factores estimulantes de colonias, manejo adecuado de las complicaciones metabólicas y apoyo de las unidades de cuidados intensivos en los casos requeridos.
3.- Fomentar talleres y/o programas educativos para padres y representantes así, como para el personal de salud comprometido con esta patología para disminuir la morbilidad infecciosa en estos pacientes.
REFERENCIAS
1. Miller KB, Daoust PR. Clinical manifestations of acute lymphoblastic leukemia. In: R. Hoffman, E. Benz, editors. Hematology: Basic Principles and Practice. 3ª ed. Philadelphia 200, pp. 999-1018. [ Links ]
2. Harrison C. Acute Lymphoblastic Leukaemia. Best Practice and Research. Clin Hematol 2004; 14(3):593-607. [ Links ]
3. IARC. Scientific publications Nº 144. International incidence of childhood cancer. Vol. II. Lyon, France 1998, pp.71-73. [ Links ]
4. Carrascal E, Collazos T. Cáncer pediátrico en área urbana de Cali, 1962-1991. Registro Poblacional de Cáncer de Cali, Departamento de Patología, Escuela de Medicina, Universidad del Valle, Cali, 1995 (Documento Técnico). [ Links ]
5. Capote L. Frecuencia del Cáncer en Venezuela. Temas Banco de Drogas Antineoplásicas BADAN 30 años. Caracas 2008; 2: 3-6. [ Links ]
6. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57-70. [ Links ]
7. Whitlock JA, Gaynon PS. Acute lymphoblastic leukemia in children. In: Wintrobes Clinical Hematology, 11th ed. Baltimore, MD 2003, pp. 1752-1774. [ Links ]
8. Pui CH, Evans WE. Treatment of Acute lymphoblastic leukemia. N Engl J Med 2006; 354:166-178. [ Links ]
9. Evans WE, Relling MV, Rodman JH, Crom WR, Boyett JM, Pui CH. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 1998; 338: 499-505. [ Links ]
10. Nachman JB, Sather HN, Sensel MG, Trigg ME, Cherlow JM, Lukens JN, et al. Augmented post-induction therapy for children with high-risk acute lymphoblastic leukemia and a slow response to initial therapy. N Engl J Med 1998; 338:1663- 1671. [ Links ]
11. Pui CH, Relling MV, Evans WE. Role of pharmacogenomics and pharmacodynamics in the treatment of acute lymphoblastic leukaemia. Best Pract Res Clin Haematol 2002; 15: 741-756. [ Links ]
12. Pui CH, Cheng C, Leung W, Rai SN, Rivera GK, Sandlund JT, et al. Extended Follow-up of Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia. N Engl J Med 2003; 349:640-649. [ Links ]
13. Nygaard U, Toft N, Schmiegelow K. Methylated metabolites of 6-mercaptopurine are associated with hepatotoxicity. Clin Pharmacol Ther 2004; 75:274-281. [ Links ]
14. Pui CH, Campana D, Pei D, Bowman P, Sandlund JT, Kaste S, et al. Treatment of Childhood Acute Lymphoblastic Leukemia Without Prophylactic Cranial Irradiation. N Engl J Med 2009; 360:2730-2741. [ Links ]
15. Pérez C. Técnicas Estadísticas con SPSS 12. Aplicaciones al análisis de datos. Pearson Educación, S.A., Madrid 2005; 15: 569-588. [ Links ]
16. Moghrabi A, Levy DE, Asselin B, Barr R, Clavell L, Hurwitz C, et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 2007; 109(3):896-904. [ Links ]
17. Chessells JM, Richards SM, Bailey CC, Lilleyman JS, Eden OB. Gender and treatment outcome in childhood lymphoblastic leukaemia: report from the MRC UKALL trials. Br J Haematol 1995; 89: 364-372. [ Links ]
18. Guevara J.M, de Insausti C, de Lara D, Arends A. Leucemia linfoblástica aguda en niños. Evaluación del protocolo nacional 80 (P.N. 80). Rev Fac Med 1988;1:58-64. [ Links ]
19. Quintero de Charry M. Resultados del tratamiento de leucemia linfoblástica aguda en niños. Rev Colomb Med 1999; 30:148-56. [ Links ]
20. López M, Alvarado M, Jiménez R, De Diego J, González C. Adolescentes con leucemia aguda linfoblástica de novo: eficacia y seguridad de un protocolo pediátrico versus uno de adultos. Gac Méd Méx 2008; 144(6):485-489. [ Links ]
21. Campbell M, Ferreiro M, Tordecilla J, Joannon P, Rizzardini C, Rodríguez N. Leucemia linfoblástica aguda. Características al diagnóstico en 100 niños. Rev Chil Pediatr 1999;7 0(4):288-293. [ Links ]
22. Baccarani M, Corbelli G, Amadori S, Drenthe-Schonk A, Willemze R, Meloni G, et al. Adolescent and adult acute lymphoblastic leukemia: prognostic features and outcome of therapy. A study of 293 patients. Blood 1982; 60(3):677-684. [ Links ]
23. Miller DR, Leikin S, Albo V, Sather H, Karon M, Hammond D. Prognostic factors and therapy in acute lymphoblastic leukemia of childhood: CCG-141: A report from childrens cancer study group. Cancer 1983; 51(6):1041-1049. [ Links ]
24. Pui CH, Relling MV, Sandlund JT, Downing JR, Campana D, Evans WE. Rationale and design of Total Therapy Study XV for newly diagnosed childhood acute lymfoblastic leukemia. Ann Hematol 2004; 83(1):124-126. [ Links ]
25. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006;354(2):166-178. [ Links ]
26. Schrappe M, Reiter A, Ludwig WD, Harbott J, Zimmermann M, Hiddemann W, et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM Study Group. Blood 2000; 95(11):3310-3322. [ Links ]
27. Perera A, Navarro C, Landolfi C, Cárdenas L, Inaty J, Insausti C, et al. Results of the long-term treatment of 209 children with acute lymphoblastic leukaemia: experience of a multicentred group in Venezuela. Clin Translat Oncol 2003;5(8): 450-457. [ Links ]
28. Pui CH, Pei D, Sandlund JT, Ribeiro RC, Rubnitz JE, Raimondi SC, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leucemia 2010; 24: 371-382. [ Links ]
29. Veerman AJ, Kamps WA, van den Berg H, van den Berg E, Bökkerink JP, Bruin MC, et al. Dexamethasone-based therapy for childhood acute lymphoblastic leukaemia: results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol 2009; 10(10):957- 966. [ Links ]
30. Gil M, Suárez M, Rivero M, Hong A. Experiencia del Servicio de Hematología del Hospital de Niños de Caracas en el tratamiento de niños con leucemia linfoblástica aguda. Arch Venez Puer Ped 2003; 66: 54-56. [ Links ]
31. Landolfi C, Perera A, Navarro C, García R, Carneiro M, de Insausti C, et al. Resultados del Protocolo 2001 del Grupo Cooperativo Nacional para el tratamiento de niños con leucemia linfoblástica aguda. Memorias del IX Congreso Venezolano de Hematología Dr. Alonso Nuñez Montiel 2007:MH27. [ Links ]
32. Urdaneta B, Montilva R, Álvarez M. Leucemias linfoblásticas agudas en pacientes pediátricos. Evaluación de la sobrevida. Año 2003 a 2007. Hospital de Especialidades Pediátricas, Maracaibo, Zulia-Venezuela. Memorias del IX Congreso Venezolano de Hematología Dr. Alonso Nuñez Montiel 2007:MH42. [ Links ]
33. Niemeyer C.M, Hitchcock-Brayan S, Sallan S.E. Comparative Analysis of treatment Programs for Childhood Acute Lymphosblastic Leukemia. Seminars in Oncology 1985; 12:122. [ Links ]
34. Campbell M, Salgado C, Quintana J, Becker A, Vargas L, Cabrera M, et al. Mejoría en el pronóstico de la leucemia linfoblástica aguda en niños de un país en desarrollo: Resultados del protocolo nacional chileno PINDA 87. Rev Chil Pediatr 1999; 70:405-414. [ Links ]
35. Buendia MT, Lozano JM, Suarez GE, Saavedra C, Guevara G. The impact of acute lymphoblastic leukemia treatment on central nervous system results in Bogota, Colombia. Pediatr Hematol Oncol 2008; 30(9):643-650. [ Links ]
36. Barry E, DeAngelo DJ, Neuberg D, Stevenson K, Loh ML, Asselin B, et al. Favorable Outcome for Adolescents With Acute Lymphoblastic Leukemia Treated on Dana Faber Cancer Institute Acute Lymphoblastic Leukemia Consortium Protocols. J Clin Oncol 2007; 25(7):813-819. [ Links ]

