










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGen
versión impresa ISSN 0016-3503versión On-line ISSN 2477-975X
Gen v.63 n.4 Caracas dic. 2009
Herencia dominante de malformación anorrectal. Estudio en una familia
Dres. Anibal Saud*, Maria Teresa Artis**, Keila Córdoba***, Jojana Guzmán****,
*Cirujano pediatra Coordinador Post grado Cirugía pediátrica,
**Gastroenterólogo pediatra,
***Residente de II año Postgrado Pediatría,
**** Residente I año Postgrado de Cirugía pediátrica. Anexo pediátrico del Hospital "Dr. Luis Razetti". Estado Anzoátegui. Venezuela. Año 2008
RESUMEN
Las Malformaciones anorrectales (MAR) son un grupo de malformaciones congénitas caracterizadas por la falta de unión entre la fosa anal y el conducto anorrectal. Afecta al varón con una frecuencia ligeramente mayor que en la mujer. Se presentan en uno de cada 4000 a 5000 recién nacidos vivos. El riesgo estimado de una pareja de tener otro hijo con una MAR es de alrededor del 1 %. Se ha descrito en algunos casos un tipo de herencia autosómica recesiva, pero la recurrencia en la hermandad es por lo general baja. Presentamos un caso de neonato masculino con diagnóstico antenatal de megacolon congénito Vs MAR quien a la exploración física se evidencia ano no permeable y se le realiza invertograma con hallazgo de MAR alta, por lo cual se realizó colostomía derivativa, y en un segundo tiempo quirúrgico se diagnostica malrotación intestinal. Resulta interesante presentarlo ya que tres miembros directos de su grupo familiar tienen antecedentes de malformación congénita del tracto gastrointestinal con variedades clínicas, siendo novedoso destacar la transmisión hereditaria de esta rara condición.
Palabras claves: malformación anorrectal, megacolon.
SUMMARY
The Malformations anorectal (MAR) are a group of congenital malformations characterized by the lack of union between the anal pit and duct anorectal. Affects men often slightly greater than in the women, and are presented in one of every 4000 to 5000 newborn. The estimated risk of a couple of having another child with a sea is about 1 %. Described in some cases by a recessive autosomal inheritance type but the recurrence in the brotherhood is generally low. We present a of newborn male with antenatal diagnosis of congenital megacolon vs. MAR physical examination is evidenced not permeable anus and is backed with invertograma with high MAR, finding which colostomy it´s why colostomy derivation itÊs mode, and at a second surgical time diagnoses intestinal malrotation. It´s made It is interesting to present it since three direct members of a family group have a history of congenital malformation of the tract gastrointestinal with clinical variety, being novel to stand out the hereditary transmission of this rare condition.
Words key: anorectal, megacolon malformation.
Recibido Sep. 2008. Revisado Nov. 2008. Aceptado May. 2009.
INTRODUCCIÓN
Estrictamente hablando, malformación congénita se define como un defecto estructural en un órgano o parte de un órgano o segmento corporal provocado por una alteración intrínseca del desarrollo, esto quiere decir una alteración en un tejido u órgano desde su origen. Los avances en el estudio de la genética han permitido conocer que todo el proceso de la morfogénesis está regulado genéticamente y se conocen actualmente una cantidad, cada vez más creciente de genes llamados genes del desarrollo que funcionan durante la vida prenatal y que son los responsables de guiar todos los procesos involucrados en el desarrollo prenatal normal. En la gran mayoría de las malformaciones congénitas (50%-60%) no es posible conocer la causa del defecto, entre 20% y 25% son de etiología multifactorial (mezcla de factores genéticos y ambientales); 7% a 10% son provocadas por agentes meramente ambientales; entre 7% y 8% son debidas a genes mutantes y entre 6% y 7% son de etiología cromosómica(1).
Las malformaciones anorrectales (MAR) representan una de las anomalías del tubo digestivo de frecuente ocurrencia, con una incidencia estimada entre 1 por 4000 a 1 por 5000 recién nacidos (RN) vivos. Existe un ligero predominio de hombres respecto de las niñas, con una relación de 1,4/1,6:1. Sin embargo, dado que la mayoría de las niñas tienen lesiones bajas, la relación entre hombres y mujeres para éstas últimas es de 1:1 y para las altas es de 1,8:1(2). Se ha descrito en algunos casos un tipo de herencia autosómica recesiva, pero la recurrencia en la hermandad es por lo general baja. Por tal motivo resulta interesante describir que el caso expuesto se presenta como una alteración de herencia autosómica dominante (cuando sólo uno de los padres le trasmite un gen anormal)(1).
Se han discutido varias causas exógenas, pero ninguna es definitiva. La definición de las anomalías anorrectales que da el International Clearinghouse for Birth Defects Monitoring Systems "es la ausencia de un ano o la falta de pasaje a través del recto, o desde el recto hacia el canal anal". También puede ocurrir que la abertura anal esté en un lugar diferente al normal (ano ectópico). El defecto puede afectar los últimos milímetros del ano o comprometer el recto, el aparato esfinteriano, la vía urogenital y las vértebras sacro-coccígeas. La frecuencia de defectos genitourinarios asociados varía de 20 a 54%. Según el nivel en donde termine el intestino (fondo de saco) y según la presencia de fístula se clasifican en malformaciones anorrectales altas y bajas. Esta anomalía se produce por alteraciones embriológicas a nivel del intestino posterior y la mayoría resultan de la división anormal de la cloaca por el tabique urorrectal, hacia recto y conducto anal en la parte posterior, y vejiga y uretra en sentido anterior. La detención del crecimiento, desviación del tabique urorrectal en sentido dorsal, o ambas, provoca la mayor parte de las anormalidades anorrectales, como atresia rectal y conexiones anormales entre recto y vejiga, uretra o vagina. Puede ser una malformación leve con fácil resolución quirúrgica, o una malformación grave, con resolución quirúrgica compleja. Entre más alta es la malformación, con mayor frecuencia se encuentran anormalidades urológicas acompañantes.
Los pacientes con cloacas persistentes o fístulas rectovesicales tienen una probabilidad de 90% de presentar un defecto genitourinario asociado. Al contrario, los niños con deformidades bajas poseen una probabilidad menor de 10% de presentar una anormalidad urológica acompañante. Los datos clínicos en los pacientes arrojan en un 80-90% de los casos el diagnósticopreciso para tomar decisiones terapéuticas(3). Las imágenes diagnósticas tienen un importante rol en el estudio de estos pacientes, que está orientado a demostrar adecuadamente la anatomía de la malformación y, por otra parte, precisar la existencia de anomalías asociadas.
El examen más utilizado inicialmente en el estudio de pacientes con malformaciones anorectales es el ultrasonido, que posteriormente se complementará con colografía distal, uretrocistografía y resonancia magnética de acuerdo a la condición clínica de cada paciente(4).
La rehabilitación del niño afecto de malformación anorrectal va a incidir en unos aspectos o en otros dependiendo de la edad. Aunque el tratamiento quirúrgico consiste en corregir el defecto anatómico colocando el recto y ano en posición normal, la funcionalidad se normaliza a edades avanzadas o incluso nunca. Por tanto el tratamiento rehabilitador debe extenderse hasta conseguir una calidad de vida aceptable, que permita al paciente desarrollar sus actividades sin restricciones(5).
El motivo de esta presentación es destacar la transmisión autosómica dominante de la malformación anorrectal en un grupo familiar, los criterios diagnósticos y la variedad clínica de esta rara condición, mostrando un caso con miembros directos de la familia con malformaciones anorrectales de diferente forma de presentación que tuvieron un seguimiento prolongado y de evolución satisfactoria en todos.
CASO CLÍNICO
Neonato masculino producto de madre de 22 años, III gesta, embarazo controlado, con reporte de serología VIH negativa y VDRL No Reactivo vigentes, Tipiaje materno grupo "A", factor Rh positivo; con diagnóstico ecográfico antenatal de malformación anorrectal Vs megacolon. Es obtenido a las 39 semanas de gestación por cesárea segmentaria electiva (por patología fetal descrita). Respiró y lloró espontáneamente al nacer; APGAR: 8 y 9 puntos; peso y talla al nacer de 3.050 gr y 50 cm respectivamente; con hallazgos al examen físico de ano no permeable.

Se ingresa en el Servicio de Neonatología siendo evaluado en conjunto con Servicio de cirugía pediátrica con hallazgos en el invertograma de malformación anorrectal alta (Figura 2), realizándose colostomía a dos bocas (Figura 3a... y 3b). En su postoperatorio mediato amerita reintervención quirúrgica por evisceración de asas delgadas; realizándose en esta oportunidad diagnóstico de malrotación intestinal. Presenta proceso infeccioso asociado y descompensación hemodinámica que se resuelve en sala de hospitalización con antibióticos de amplio espectro y manejo médico; y por evolución satisfactoria es egresado a los 14 días con control por consulta externa.


ALTERACIONES EN OTROS MIEMBROS DE LA FAMILIA
Padre: 34 años de edad. Ameritó anoplastia por malformación anorrectal tipo membrana anal. Hermano paterno: Varón de 10 años de edad (Figura 4), quien a los 4 años ameritó corrección quirúrgica de hernia diafragmática, con hallazgos intraoperatorios de megacolon a nivel de sigmoides; (no se le realizó toma de muestra para Biopsia ni estudios pertinentes). Actualmente estreñimiento crónico tratado con laxantes y dieta rica en fibras.

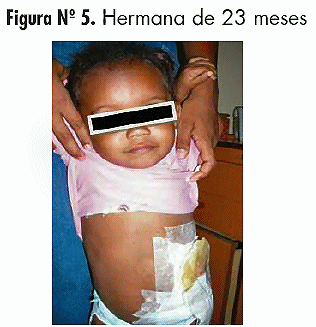
Hermanas: Dos hembras de la misma unión conyugal (Figura 6); una de 3 años con estreñimiento crónico, actualmente en tratamiento dietético y laxantes; y una de 23 meses (Figura 5) quien a partir del sexto mes de vida comienza a presentar estreñimiento severo con distensión, dolor abdominal y vómitos a repetición, siendo controlada con gastroenterólogo pediatra quien realiza colon por enema con evidencia de segmento de colon dilatado seguido de zona de transición a nivel de colon sigmoides, realizándose diagnóstico de Megacolon congénito. A los 8 meses presenta cuadro obstructivo intestinal bajo que ameritó Laparotomía exploradora, con hallazgos preoperatorios de estenosis rectal, ano anterior y fístula rectovaginal e intraoperatorios de megacolon (1er tiempo). Se toma de muestra para biopsia de la zona de transición, que concluye: Aganglionosis a nivel de capa muscular del segmento. A los 21 meses se le realiza anorrectoplastia posterior más cierre de fístula recto vaginal (2do tiempo).

DISCUSIÓN
En el transcurso de las últimas 4 décadas, el diagnóstico prenatal ha mostrado un desarrollo impresionante y rápido, tanto en el número de mujeres estudiadas como en problemas y trastornos, para los que se cuenta con las pruebas idóneas. Más de la mitad de los casos pueden ser identificados en etapa prenatal, ya que la mayoría de los hallazgos son sospechados en la ultrasonografía inicial(6).
El diagnóstico prenatal acertado de las malformaciones congénitas y enfermedades genéticas y su posterior atención, contribuyen a mejorar los indicadores de mortalidad fetal, perinatal e infantil. Los estudios prenatales permiten detectar anomalías estructurales o cromosómicas y descartar una gran cantidad de defectos congénitos genéticos cuya presencia puede sospecharse debido a los antecedentes familiares(7). En nuestro caso el diagnóstico prenatal por ecografía de malformación anorrectal y los antecedentes familiares fueron cruciales para el manejo posterior al nacimiento y el consejo genético.
Se ha descrito que el riesgo estimado de una pareja de tener otro hijo con una malformación anorrectal es alrededor del 1 % y en algunos casos se ha descrito un tipo de herencia autosómica recesiva(5). En este grupo familiar consideramos una forma de transmisión de la enfermedad anorrectal de tipo autosómica dominante, ya que fue transmitido del padre a hijos de diferente unión conyugal.
En un estudio publicado por Falcone y colaboradores encontraron en 1606 pacientes con malformaciones anorectales, que 39 de ellos (2,4%) tenían un miembro de la familia con anomalías congénitas. Encontraron anomalías congénitas no asociadas a malformaciones anorectales como masas sacras y anomalías ginecológicas, hematológica, del esófago, duodenales, renales, y espinales; 24 pacientes (1.4%) tenían 1 ó más miembros de la familia con una malformación anorectal. Entre las hembras con antecedentes familiares positivos, el 73% hicieron una fístula vestibular o perineal, comparado con sólo un 36% en pacientes sin antecedentes familiares. Entre varones, el 35% tenían fístulas perineales comparadas sólo un 10% de ésos sin miembros afectados de la familia. Ellos concluyen que una historia familiar positiva en 1,4% representa un fuerte componente genético para MAR. Estos nuevos datos permiten un asesoramiento más informado de familias con una malformación anorectal y apoyan la necesidad de otros estudios genéticos(6).
No han sido publicados casos similares en Venezuela ni a nivel mundial, por lo que se considera el primer caso de malformación anorrectal familiar de transmisión autosómica dominante. La asociación de malformación anorrectal y enfermedad de Hirschsprung es sumamente rara, resultando un reto diagnóstico(8), siendo interesante la descripción del caso confirmado por biopsia de una de las hermanas y el no confirmado del hermano paterno.
Algunos antecedentes maternos demostraron ser importantes factores de riesgo, como metrorragia del primer trimestre y otros miembros afectados en la familia que se encontraron casi en la cuarta parte de los casos, reportan Julio Nazer y Col(3). En nuestro caso no hubo antecedentes maternos positivos.
En todos estos pacientes es necesario efectuar cariograma, no sólo para diagnosticar el sexo, sino también para descartar anormalidades cromosómicas y realizar un buen consejo genético. Es también muy importante realizar estudio radiológico de columna lumbosacra e incluso resonancia magnética, con el objeto de completar el estudio, ya que las malformaciones espinales son un hallazgo frecuente en pacientes con anomalías anorrectales(3).
Se concluye que un buen diagnóstico prenatal, unido a un asesoramiento genético correcto y a una adecuada atención multidisciplinaria, constituye una forma de medicina preventiva que abre nuevos horizontes, ayuda a disminuir la ansiedad familiar y asegura que las personas con alto riesgo puedan ejercer el derecho a la reproducción de manera informada.
REFERENCIAS BIBLIOGRAFICAS
1. Bojorge, E. Prevalencia y factores asociados a los defectos congénitos en el Servicio de Neonatología del Hospital Fernando Vélez Paiz, 01 de Enero al 31 de Diciembre del 2003. Managua-Nicaragua, 2004. Disponible en la World Wide Web: http://www.minsa.gob.ni/bns/monografias/Full_text/Pediatria/servicio_neonatalogia.PDF. [ Links ]
2. Arango M., Múnera A., Manotas R. Experiencia en el Hospital Universitario San Vicente de Paúl sobre el manejo quirúrgico de los pacientes con ano imperforado. Revista colombiana de cirugía. 2005. Disponible en la World Wide Web: http://www.encolombia.com/medicina/cirugia/Ciru20105-Experiencia.htm. [ Links ]
3. Nazer J., Hubner M., Valenzuela P. et al. Malformaciones congénitas anorrectales y sus asociaciones preferentes. Experiencia del Hospital Clínico de la Universidad de Chile. Período 1979-1999. Rev. méd. Chile 2000,128 (5). Disponible en la World Wide Web: http://www.scielo.cl/scielo.php?pid=s0034-98872000000500010&script=sci_arttext.
4. Moënne, K. Imágenes en anomalías anorectales. Rev Chil Radiol 2003; 9: 13-18. Disponible en la World Wide Web: http://www.imagenologia.cl/rad91/moenne.html. [ Links ]
5. Panzuto O., Bellia P., Millán F. et al. Malformaciones anorrectales. Disponible en la World Wide Web: www.elizalde.gov.ar/area_medica/Normas/.../maflorm_ano.doc. [ Links ]
6. Falcone Jr., Levitt MA., Peña A., et al. Increased heritability of certain types of anorectal malformations. J Pediatr Surg. 2007 Jan;42(1):124-7;discussion 127-8. Disponible en la World Wide Web: http://www.ncbi.nlm.nih.gov/pubmed/17208552. [ Links ]
7. Piloto M., Sanabria M., Menéndez R. Diagnóstico prenatal y atención de las malformaciones congénitas y otras enfermedades genéticas. Rev Cubana Obstet Ginecol 2001; 27(3):233-40. Disponible en la World Wide Web: http://bvs.sld.cu/revistas/gin/vol27_3_01/gin11301.htm. [ Links ]
8. Alvarado R., Jiménez P., Gallego J. Agangliosis del colon en pacientes con malformación anorrectal, análisis de cinco casos. Cirugía y cirujanos. Año/vol. 73, No 004. Academia mexicana de cirugía. Distrito Federal, México. 2005. pp. 283-286. Disponible en la World Wide Web: http://redalyc.uaemex.mx/redalyc/pdf/662/66273407.pdf. [ Links ]

