










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Latinoamericana de Metalurgia y Materiales
versión impresa ISSN 0255-6952
Rev. LatinAm. Metal. Mater. v.30 n.1 Caracas jun. 2010
Effects of gamma radiation in polymer blends, in composites with micro and nano fillers and in functionalized polyolefins
Carmen Albano 1,2*, Rosestela Perera 3, Pedro Silva 4
1: Instituto Venezolano de Investigaciones Científicas, Centro de Química, Caracas, Venezuela.
2: Universidad Central de Venezuela, Facultad de Ingeniería. Caracas, Venezuela.
3: Departamento de Mecánica, Universidad Simón Bolívar. Caracas, Venezuela.
4: Instituto Venezolano de Investigaciones Científicas, Centro de Física. Caracas, Venezuela
E-mail: carmen.albano@ucv.ve
Publicado On-Line el 14-Jun-2010
Disponible en: www.rlmm.org
Resumen
La radiación gamma se utiliza frecuentemente para la esterilización de algunos artículos médicos hechos de plástico, pues se sabe que descompone las moléculas de ADN de los organismos vivos. Sin embargo, este tipo de radiación puede producir cambios en la estructura química de los polímeros, que resultan en modificaciones de sus propiedades macroscópicas. En esta revisión se presentan algunos trabajos publicados por nuestro grupo de investigación en el área de radiación gamma aplicada a polímeros termoplásticos como polietilenos y polipropilenos; a compuestos de poliolefinas con cargas orgánicas como la fibra de sisal y el aserrín, o con hidroxiapatita y residuos marinos para aplicaciones biomédicas y los cambios que resultan en su comportamiento mecánico y térmico. Otros tópicos incluidos en este trabajo son los efectos producidos por la irradiación de mezclas de polímeros con cauchos termoplásticos como el copolímero estireno-butileno-estireno (SBS); de mezclas de polipropileno y poliestireno, o de polietileno con poliamida 6 y con otras poliolefinas. Adicionalmente, se describirán algunas investigaciones donde se usó la radiación gamma para inducir la funcionalización de poliolefinas.
Palabras Claves: Radiación Gamma, poliolefinas, mezclas, compuestos, comportamiento mecánico y térmico
Abstract
Gamma radiation is commonly used in the sterilization of some medical articles made of plastics, because it decomposes the DNA molecules of living organisms. Nonetheless, this type of radiation can produce changes in the molecular structure of polymers, which result in changes in their macroscopic properties. In this review, some published works of our group regarding the changes in the mechanical and thermal behavior produced by irradiation of thermoplastics such as polyethylenes and polypropylenes; of composites of polyolefins with organic fillers such as sisal fiber and woodflour, or with hydroxyapatite and marine residues for biomedical applications, will be presented. Other topics included in this review are the effects produced by irradiation in blends of polymers with thermoplastic elastomers such as styrene-butylene-styrene (SBS); of blends of polypropylene with polystyrene, or polyethylene with polyamide 6 and with other polyolefins. Additionally, some works where gamma radiation was used to induce the functionalization of polyolefins will be described.
Keywords: Gamma radiation, polyolefins, blends, composites, mechanical and thermal behavior
Recibido: 25-Jul-2009; Revisado: 01-Ene-2010; Aceptado: 17-Feb-2010
Contents
1.-Introduction
2.-Mechanical Behavior of Irradiated Polymers, Blends and Composites
2.1- Irradiated Polymers
2.2- Irradiated Blends and Composites
2.3- Conclusions
3.-Thermal Behavior of Irradiated Polymers, Blends and Composites
3.1- Irradiated Polymers
3.2- Irradiated Blends and Composites
3.3- Conclusions
4.-References
1. INTRODUCTION
The use of radiation in polymer processing is gaining more and more interest because it can be suggested as an alternative to traditional chemical methods to modify their molecular structure [1]. Ionizing radiation induces chemical reactions in polymers, which result in changes in both the molecular structure and macroscopic properties [2- 4]. The possibility of processing the final shape of the polymeric material in the solid state opens up new opportunities to obtain materials with welltailored properties.
On the other hand, ionizing radiation, specifically gamma radiation, is one of the methods most commonly used for sterilization purposes, because it decomposes the DNA molecules in living organisms and can be applied simultaneously to sterilize many products with low energy consumption. Gamma irradiation allows the sterilized materials remain as such for long periods of time.
A large number of medical supplies that need to be sterilized are made of polymers. Hence, detailed studies on the effects of g-irradiation onto plastic materials are needed to increase the variety of polymer blends and composites suitable for specific purposes.
Among the published works of our group in the field of gamma radiation applied to polymers are those regarding the mechanical and thermal behavior of thermoplastics such as polyethylenes [5] and polypropylenes [6]; of composites of polyolefins with organic fillers such as sisal fiber and woodflour [4,7], or with hydroxyapatite and marine residues [8-11] for biomedical applications. Other studies include the radiation effects on blends of polymers with thermoplastic elastomers such as styrenebutylene- styrene (SBS) [12]; of blends of polyproylene with polystyrene [13-15] or polyethylene with polyamide 6 [16] and with other polyolefins [17]. Some others report the use of gamma radiation to induce the functionalization of polyolefins [18]. In this review, some of such findings will be presented.
2. MECHANICAL BEHAVIOR OF IRRADIATED POLYMERS, BLENDS AND COMPOSITES
2.1 Irradiated Polymers
The effects of ionizing radiation on polymers include: i) crosslinking and/or chain scission, ii) formation of small molecular fragments, and iii) modification of the molecular structure. Molecular weight changes resulting from (i) can markedly influence the material properties [19-21]. A competition between scission and crosslinking reactions has been noticed in some irradiated polymers. The prevailing effect brings about macroscopic changes in the molecular mass distributions, continuously affecting transition temperatures, diffusion coefficients and mechanical properties after polymer irradiation.
Polyethylene (PE) is one of the most widely used thermoplastic polymers. For some applications, its chain structure must be modified. Commercially, both linear and branched polyethylenes may be used in applications requiring crosslinked materials provided that the chain structure is modified by Gamma irradiation. Cataño et al. [5] studied the changes in mechanical behavior of gamma irradiated high-density, low-density and linear lowdensity polyethylenes (HDPE, LDPE and LLDPE) with storage time (ts) in air, at integral or absorbed doses (Di) between 15 and 100 kGy and 4.8 kGy/h of dose rate, in air, at room temperature. An initial decrease in tensile strength at the different radiation doses was reported with storage time, which eventually leveled off. As an example, a low-density polyethylene displayed a tensile strength drop from 21.6 MPa in the non-irradiated condition to values between 11.0 and 13.3 MPa after irradiation at the different doses. The same trend was observed when the elongation at break was evaluated: samples become brittle when the storage time was increased, and fracture followed immediately after the yield point. These values remained almost constant with storage time in an interval of 320 days, which implies that changes in the polyolefins structure are being produced mostly just after irradiation and more extensively in the amorphous phase. The initial drop, once the samples were exposed to radiation, is the result of a drawability loss, as a consequence of crosslinking reactions that were produced by radiation. This effect was evidenced through gel content determinations, which varied between 70 and 90% in all the PEs tested in that research. The degradative processes become slower with the storage time, which could be ascertained from the total free-radical concentration measurements in these polyolefins in their nonirradiated, irradiated and after one-month storage conditions [22] (Figure 1).

Kusumoto et al.[23] indicate that radical decay in crystalline polymers, especially in polyethylenes, involve two processes: one rapid at the beginning, followed by a slower one. According to Dole [19-20], the rapid decay in the radicals occurs in the non-crystalline phase and generally takes place at the beginning of the experiment. The slower process, on the contrary, is attributed to the diffusion of the radicals from inside the crystals to their surface where they readily react.
2.2 Irradiated Blends and Composites
Fillers are added to thermoplastics for two main purposes: to increase the volume of the final product to reduce the cost of the materials, on the one hand, and to change their mechanical properties, on the other. These changes are frequently far from reaching theoretically predicted values.
The quality of the filler depends on many factors such as the average particle size and its distribution, their shape and porosity, the chemical nature of their surface, etc. Examples of some commonly-used organic fillers are sisal fibers, woodflour, cotton, etc. Some inorganic fillers also commonly used are CaCO3, talc, montmorillonite, hydroxyapatite, etc.
Cellulose fibers are excellent reinforcing elements for the preparation of polymer composite materials. Given their elastic nature, they can be more easily processed than some more rigid fibers such as fiberglass [24-25]. Disadvantages associated with the use of natural fibers as reinforcement in thermoplastics are the result of a lack of a good interfacial adhesion and a poor resistance to humidity absorption, since cellulose fibers are of hydrophilic nature and thermoplastics are usually hydrophobic [26].
The irradiation of composites could be an alternative to coupling or compatibilizing agents to promote some interactions between the phases. Its use has been studied to improve the filler/matrix interface, in a simpler, cleaner and cheaper way. This is the only technique that introduces energy into a material to generate favorable changes in its structure, provided it is used in the proper doses and under the appropriate conditions.
The tensile behavior (tensile strength, elongation at break) and impact strength of polypropylene (PP) filled with sisal fiber (20 wt.%) and woodflour (40 wt.%) irradiated at low integral doses (0-70 kGy) and at a dose rate of 4.8 kGy/h in air at room temperature with a 60Co source are displayed in figures 2-4 [4]. The trends in these properties are similar for both types of composites studied, but values found for the PP/sisal fiber composites are higher than those for PP with woodflour.
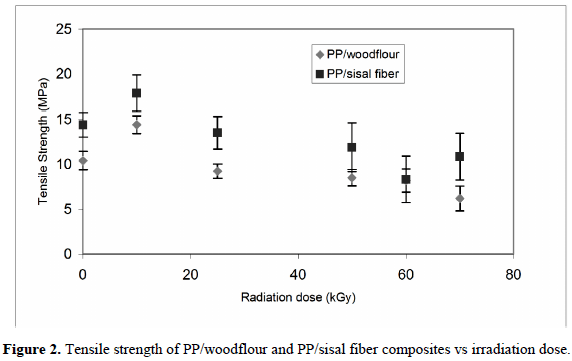


Tensile strength and elongation at break increase 38 and 35% in the PP/woodflour composites and 24 and 25% in the PP/sisal fiber composites, respectively (figures 2 and 3), when the radiation dose was 10 kGy. The change of these mechanical properties at low irradiation doses (10 kGy) can be attributed to several reasons: first, to a slight crosslinking which may be experienced by the PP [27-32], and second, to a higher polymer-filler interaction due to the increase in the hydrophilicity of PP after irradiation, owing to an increase in the concentration of functional OH groups [33]. The fillers have also high concentrations of OH groups which may promote interactions such as hydrogen bonding between them and the PP. These interactions can be estimated through a wide band within 3300 and 3400 cm-1 on the infrared spectrum of PP. In summary, surface oxidation of PP increased its adhesion to the organic fillers.
With the increase of the radiation dose, degradation through the formation of peroxy and hydroperoxy radicals in the amorphous region appears to be the primary cause of physical property losses. Electronic Paramagnetic Resonance (EPR) was used by Albano et al. [4] to study the radical concentration in samples of PP/sisal fiber and PP/woodflour after irradiation. The spectra showed that the formation of peroxy radicals strongly increases with the radiation dose, being higher in those samples with woodflour (figure 4). This behavior unfolds that the main degradation mechanism in these PP composites is through chain scissions, which strongly influences their mechanical, thermal and morphological behavior. When enough numbers of tie molecules between crystallites are cut through this chain scission process, the local stress concentrations on the crystals increase. Therefore, mechanical properties such as tensile strength and elongation at break are reduced drastically (figures 2 and 3).
Impact strength (figure 5) of PP/sisal fiber and PP/woodflour composites decreases as a function of the radiation dose. The decreases after irradiation at 10 kGy are 24% and 44%, respectively. At higher irradiation doses, the impact strengths further decrease to 35 and 9% of the initial values, respectively. Micrographs of the fractured surface of the PP/woodflour and PP/sisal fiber samples (figure 6) indicate that the fillers were not optimally dispersed and tend to impair the mechanical properties of the composites.


Albano et al. [7] analyzed the effect of different integral doses (0-70 kGy) of g irradiation from a 60Co source at 4.8 kGy/h in air and at room temperature, on the mechanical properties of blends of PP with virgin and recycled HDPE (v-HDPE, r- HDPE, respectively) filled with 40 wt.% of woodflour, by means of simple mathematical models. From their results, they concluded that the resultant properties are a function of the integral dose. Dependences of the Youngs modulus and elongation at break on the integral doses were taken for a given constant dose rate, to explain the type of processes occurring at a particular irradiation dose.
According to Chipara et al. [34], degradation and cross-linking during irradiation can be represented by means of the following processes:
A) A first order (monomolecular) process, which is equivalent to an exponential dependence of the property (Y) on Di:
Y=Po exp (-KDi)=aYo exp(-KDi) (1)
where Yo is the property of the non-irradiated sample, a a pre-exponential factor and K the rate constant associated to the monomolecular decay of the property. The deviation of a from unity is a measure of the non-linear contribution to the degradation process.
Due to the chemical decomposition of the samples, the dependence of Y on Di may be described as a superposition of two processes:
Y=Yo1 exp (-K1Di)+Yo2 exp(-K2Di) (2)
where K1, K2 represent the rate constants, a1, a2 are the pre-exponential factors, Po1=a1Yo1 and Po2=a2Yo2. This expression (2) may also describe the inhomogeneous degradation.
B) A second order (bimolecular) dependence of the property on the integral dose:
Y=Yo+ADi2 (3)
where A is a constant for a given dose rate.
The possibility of two competing degradation processes has to be considered:
Y=Yo + A1 Di2 + A2 Di2 or Y= Yo +A1Di2 +A2 Di3 (4)
These relatively simple expressions are capable of describing or indicating the type of process taking place when the composite is submitted to the effects of irradiation.
Figures 7 and 8 show the experimental values and the mathematical analysis of the Youngs modulus of composites of PP/v-HDPE/woodflour and PP/r- HDPE/woodflour.


An initial decaying behavior of the Youngs modulus is observed, which continues up to a dose of 50 kGy for the PP/v-HDPE/woodflour composite, and up to 30 kGy for the PP/r-HDPE/woodflour one. In the case of the composites with recycled polyethylene, the following increase in modulus takes place at lower irradiation doses due to the characteristics of the recycled material (r-HDPE), because it presents previous degradation and/or crosslinking.
The behavior of Youngs modulus for these composites is somehow similar to that for the PP/HDPE blends [7], due to the influence of HDPE. The initial decay of the modulus (at low absorbed doses) can be explained based on the destruction of the crystallites, which act as physical crosslinks, holding the polymer chains together. Such physical crosslinks are not real chemical bonds, because they disappear when the polymer melts. These kinds of crosslinks have a stronger effect on the modulus than those produced via a chemical reaction; whereas at higher radiation doses, the increase in the chemical crosslinking density compensates for the further loss of crystallinity and leads to an increase in the modulus. The behavior of the elastic modulus is very similar to that of the density. Initially, a decrease in density is observed, corresponding to the destruction of the crystalline region in the polymer. When almost all crystals have been destroyed, density rises again as a result of the structural changes in the polymer, i.e. the formation of the double bonds and crosslinks, which bind the molecules more closely together and lead to a tighter packing of the polymer chains. Chapiro [30] also explains that the elastic modulus of HDPE, at room temperature, decreases at low doses, but then it increases again at higher integral doses. This explains the behavior of the PP/HDPE blends, where the predominant influence of HDPE on the studied polyolefin blends is ascertained. Moreover, the effect of this polymer on the Youngs modulus values is stronger than that of a rigid filler such as woodflour at lower irradiation doses.
The results for Youngs modulus calculations using the mathematical models obtained for both composites in the 0-50 kGy range are as follows:
a)PP/v-HDPE/wood:
E=-14Di3 + 106Di2 - 208Di + 1135 (5)
b)PP/r-HDPE/wood:
E=+738e-0.4Di+396e-0.12Di+199e+0.32Di (6)
In this analysis of the mechanical properties of the PP/v-HDPE/woodflour composite, the cubic polynomial equation obtained points to a more complex dependence of the Youngs modulus on the integral irradiation dose. Not only the polymers have influence on this composite, but also the filler characteristics and the filler/matrix adhesion must be taken into consideration.
The PP/r-HDPE/woodflour composite displays a complex behavior, but the equation can be reduced to the following: E=+396e-0.12Di+199e+0.32Di , which indicates that the crosslinking and degradation (chain scission) processes are competitive or overlapped. This expression can also mean an inhomogeneous degradation. These variations in the mathematical analysis, which are observed when the mechanical property of both composites are analyzed, can be explained based not only on the presence of the wood, but on the previous degradation reactions existing in r-HDPE and those arising with the irradiation in the recycled material. In summary, although this composite shows different dependencies of the mechanical parameter on the irradiation dose, both expressions indicate simultaneous reticulation and degradation processes.
The following equations are obtained for the range within 50 and 70 kGy:
a)PP/v-HDPE/wood:
E= -176Di2 + 2182Di - 5518 (7)
b)PP/r-HDPE/wood:
E= +54.4 Di2 - 752Di + 3687 (8)
The equations used to calculate the Youngs modulus follow a bimolecular process for both composites at the above mentioned doses; this means that this mechanical parameter shows a second order dependence on the integral radiation dose, due to the presence of the wood.
As already mentioned, irradiation is also employed to sterilize composite materials used in the biomedical field. Therefore, it is important to study the changes introduced by the irradiation process onto polymeric materials such as composites of high-density polyethylene and hydroxyapatite (HA) intended for biomedical applications. Albano et al. [8] analyzed such composites prepared via solution using decalin, with different concentrations of hydroxyapatite (varying from 10 to 30 phr) after being exposed to gamma radiation at absorbed doses between 25 and 100 kGy, at a dose rate of 4.8 kGy/h.
Table 1 displays the Youngs modulus values as a function of the concentration of HA for each one of the absorbed doses. A slight increase in this tensile parameter at 0 kGy is noticed when HA content increase, evidencing a reinforcing effect exerted by the filler. When the radiation effect was studied, it was noticed that radiation did not modify the composites Youngs Modulus in a significant way. This phenomenon was also reported by Dole [19], who explained that radiation effects in doses up to 1000 kGy are relatively unimportant, because Youngs modulus and yield stress are strongly dependent on the crystallinity degree and radiationinduced crosslinks or chain scissions are minor perturbations. Only at very high levels of radiation (over 2000 kGy) these effects become important, according to Dole [19].

Changes in the tensile strength of the composites with the absorbed dose and filler content are reported in figure 9. As it can be seen, the addition of HA increases this mechanical parameter at all the absorbed doses. For each absorbed dose, the toughness of the polymer and the rigidity of the reinforcing filler (HA) are combined, thus rendering an improvement in the mechanical properties of the composites [35]. In general, the tensile strength increases with the addition of the HA in concentrations up to 20 phr. At 30 phr of the filler, the average tensile strength is lower than that of the composites with 10 and 20 phr of filler, probably due to the presence of agglomerates which act as defects that may initiate cracks and hence the rupture of the material [36]. In the same figure, it is also seen that the effects of the absorbed doses are not significant on the strength of the different composites, which implies that in this case, the filler content and the crystallinity degree of the composite prevail determining this mechanical parameter.

Some theoretical models were used to analyze the tensile data. The Youngs modulus was predicted using the Guth-Smallwoods equation [37-38]:

where Ec is the Youngs modulus of the composite (MPa), Em is the Youngs modulus of the matrix (MPa) and ff is the filler volume fraction.
Furthermore, to predict the theoretical value of the tensile strength, the equation of Nicolais-Narkis [39] was employed:
![]()
where sc is the composites tensile strength (MPa) and sm is the matrix tensile strength (MPa).
Tables 2, 3 and 4 display the dependence of the Youngs modulus, tensile strength and elongation at break values of the composites (Ec, sc, ec) normalized to the respective values of the Youngs modulus, tensile strength and elongation at break values of the matrix, on the weight fraction of the filler and on the absorbed dose.



From the results presented in Table 2, it can be concluded that the values of Ec/Em of the unirradiated composites obtained through equation (9) increase with the HA content, indicating that the incorporation of HA increases the rigidity of the HDPE, through the restriction in the mobility of the polymer molecules. On the other hand, the radiation of the composites has a random influence on these results, that is, they do not follow a certain trend. The radiation process produced a predominance of chain scission reactions in these composites obtained via solution, which, along with the presence of agglomerates at filler contents above 20 phr and the HDPE high crystallinity degree, strongly determined the mechanical behavior of these materials. For this reason, the theoretically values predicted using the equation of Guth-Smallwood are not well correlated to the experimental data, owing to the fact that some assumptions were made by those authors before proposing such an equation, which do not apply in this work. They are: the equation takes into account that the filler must be evenly distributed, there must not be filler-filler interactions and the particles must be rigid and spherical. These assumptions are not exactly true in this case, because HA particles are neither spherical nor rigid and tend to agglomerate themselves when in high proportions. Furthermore, filler-filler interactions could be possible in these composites when the filler content increases.
The tensile strength of the composites was analyzed using the theoretical model of Nicolais-Narkis, in order to understand the generation of discontinuities or weak points in the structure of theses two-phase systems. The predicted theoretical values of the ratio sc/sm are smaller than the experimental ones in all the range of HA contents and radiation doses, though there are a few exceptions. The Nicolais-Narkis equation describes structures where the adhesion is poor, because the weightage factor (1.21) is believed to be dependent on the adhesion quality between the matrix and the inclusion. A value of 1.21 of the weightage factor is stated to be valid for the extreme case of poor adhesion and spherical inclusions [40]. Equation (10) was followed using a factor P instead of the value of 1.21, being the values of P negative as seen in Table 5, because the composites tensile strength is higher than that of the matrix. According to Maiti and Lopez [40], the polymer-filler adhesion improves as the P values decrease. Hence, it can be concluded that a good interfacial adhesion is present in HDPE/HA composites prepared via solution. This fact was confirmed when the Kunori and Geils equation [41] was used. This equation relates the tensile strength with a proportionality parameter a, which is a stress concentration parameter. A higher value of a corresponds to a stronger stress concentration. The Kunori & Geils equation is as follows:
![]()

The values of a are shown in Table 6. As it can be seen, those values are negative, which leads to the same conclusion as before, that is, there seems to be a good interfacial adhesion in the composites. Tables 5 and 6 display that the values of P and a increase when the composites are irradiated at 100 kGy of absorbed dose. This fact could be attributed to a slight hindrance of the polymer-filler interaction.

Composites of polypropylene with HA behave differently [11], that is, the tensile strength (sR) (figure 10a) decreases with the addition of HA in the irradiated and non-irradiated samples, due to the presence of large agglomerates that form defects which act as crack initiation sites. The elongation at break (eR) (figure 10b) shows a drastic decrease when the filler is added to the PP, and then a small but continuous decrease with higher HA additions. This is attributed to the formation of large agglomerates with poor interfacial adhesion. This effect has also been reported to result in early fracture of the polymer matrix [42-43].

On the other hand, figure 10a shows that the radiation dose does not have a predominant effect on the tensile strength of these materials. However, a continuous decrease in their elongation at break (figure 10b) with the radiation dose is observed. This behavior is attributed to the poor interfacial adhesion between the HA and the PP and to the degradation reactions produced in the PP by g-radiation. Lack of adhesion between the two phases results in early failure at the polymer-HA interface.
The effect of g-radiation on composites of poly(methyl methacrylate) (PMMA) or high-density polyethylene with seaweed residues (SR) in the 0-100 kGy dose range at a dose rate of 4.8 kGy/h has also been studied [9-10]. Figure 11a illustrates the yield stress and tensile strength of the HDPE/SR composites as a function of the filler content and radiation dose. The yield stress decreases with filler content and tends to increase slightly with radiation dose. The tensile strength tends to increase with the radiation dose at the higher filler contents. At the lowest concentration of the filler (20%), however, this does not apply. The yield strain also decreases with the inclusion of the filler but remains constant with the radiation dose (figure 11b). The yield properties are controlled by debonding-yielding processes. First, the load is distributed throughout the filler/matrix interface and afterwards, the yielding of the matrix follows. Therefore, particulate composites are less elastic due to the agglomerates which may act as weak points of stress concentration on composites, producing lower yield points. The higher the filler content is, the lower the yielding of the matrix will be. Elongation at break decreases when the filler content and radiation dose increase. This fact is attributed to chain scissions in the amorphous region (figure 11b).

According to Chapiro [30], g-radiation has two effects in polymers: chain branching and/or crosslinking and chain scissions. Both processes can take place simultaneously, and which one prevails depends on the polymer nature, the surrounding atmosphere during the irradiation and the integral dose. The degradation is easily evidenced in PMMA through polymer discoloration even at low absorbed doses. A low value of gel content (2.5% ) at 100 kGy of integral dose was obtained, indicating the predominance of the chain scission mechanism of degradation.
An increase in Young's modulus is observed when seaweed residues are incorporated into the PMMA matrix (Table 7). Nonetheless, radiation does not seem to modify the modulus values at the applied integral doses.

Being the PMMA a polymer that tends to undergo degradation by Gamma radiation, some authors have established that the tensile strength is the mechanical property more affected by it. Drops of up to 50% in tensile strength in PMMA irradiated at an integral dose of 650 kGy have been reported [30,44]. The same trend was observed by Albano et al. [9] and Cataño et al. [10] in pure PMMA (Table 8). However, when seaweed residues were added to PMMA, a slight stabilizing effect was obtained. The elongation at break decreased in these composites with the addition of the filler and with the irradiation.

Some other studies regarding the effects of gamma radiation on polymer blends have been carried out. Albano et al. [12-14] analyzed blends of polystyrene (PS) with polypropylene (PS/PP, 80/20) with 7.5 wt.% of a styrene-buthylene-styrene triblock copolymer (SBS) added as compatibilizing agent. The study of the irradiation chemistry of PS is of particular interest since the molecular structure of this polymer contains large numbers of benzene rings, which are known as protective actions in many radical-chemical processes. Indeed, PS is found to be one of the most stable polymers with respect to radiation and very large doses are required to produce any noticeable change. Furthermore, when the room temperature (and irradiation temperature) is below the polymers Tg (which is the case in PS), almost no degradation is noticed. When the radiation is applied at T<Tg, the segmental motions are frozen and the degradation is negligible. Therefore, its behavior in the blends should be similar to its behavior when it is not mixed with any other polymer.
The impact strength (IS) of pure PP decreased significantly after irradiation (Table 9). On the contrary, PS and its blends of PS/PP with and without the compatibilizer did not show significant variations with irradiation at the integral dose of 70 kGy. Furthermore, the blend of PS/PP with SBS showed higher IS values than those of the blend without it, attributed to the elastomeric nature of SBS, the stronger interaction at the interface and the smaller size of the dispersed phase, as stated by Hlavata et al. [45] in their studies.

On the other hand, properties at the break point of blends of PS/PP without compatibilizer (Table 9) showed that non significant changes take place at the irradiation dose of 70 kGy, whereas for the same blends with SBS, the trend of these tensile properties (sR and eR) is to a slight decrease. In the blends without compatibilizer, this behavior is attributed to the PS benzene ring, which is in high concentration in the blend and serves as a protector and retardant of the PP decomposition process within this irradiation range (0-70 kGy). Another explanation would be that PS merely serves as a diluent to irradiation in blends, rather than absorbing radiation from the PP chains as is observed in the PS/PP blend. In the case of the PS/PP blends compatibilized with block SBS, the slightly decreasing trend of the properties at the break point is the result of the crosslinking and cyclization processes, generated by the SBS polybutadiene block due to its irradiation, which bring about an increase in the copolymer molecular weight.
Table 10 displays that the IS of the compatibilized and non-compatibilized blend samples irradiated at high absorbed doses (70-1300 kGy) present the same decreasing behavior as the neat polymers when the dose is increased, but in a more drastic fashion, that is, variations went from 2.5 J/m and 13.8 J/m at 70 kGy to <1.0 J/m and 1.8 J/m at 1300 kGy, respectively.

The behavior of the elongation at break and tensile strength of the PS/PP blend without compatibilizer (Tables 11 and 12) is similar to that of PS, whereas values of these properties for the compatibilized PS/PP blend show a decreasing behavior. Studies conducted by Vishwa Prasad and Singh [46] indicate that a phase separation (styrene and butadiene) takes places in SBS due to degradation, forming microcracks that result in a deterioration of its mechanical behavior. Schnabel et al. [47] demonstrated that at high irradiation doses (>1000 kGy) intermolecular crosslinks were generated in SBS, whereas at low doses, unsaturations are produced in the butadiene block of the copolymer. Dole [19], in turn, reported that at very high irradiation doses, styrene-butadiene-based copolymers showed crosslinking, hardening and embrittlement of the rubber phase, thus resulting in a sudden decrease in mechanical properties.


The incorporation of irradiated SBS into PP to modify its tensile properties was studied by González et al. [12], who reported decreases in the Youngs modulus and significant increases in the elongation at break of the blends, even with SBS irradiated at the highest absorbed dose tested of 50 kGy (Table 13). They also concluded that it is possible to vulcanize the SBS without the need of additives which are generally toxic, and at the same time, to obtain products that could be employed in applications where innocuity is required, like food packaging.

2.3 Conclusions
From the results presented here, it can be concluded that in general, the radiation affects the mechanical properties of pure polymers, their blends and composites, due to the generation of free radicals and their decay during storage of the samples. This implies that chain scission, chain branching and crosslinking are taking place. These results were confirmed through theoretical predictions of the mechanical properties.
3. THERMAL BEHAVIOR OF IRRADIATED POLYMERS, BLENDS AND COMPOSITES
3.1 Irradiated Polymers
It is well known that the main effect of the interactions of gamma rays with polymers is the formation of free radicals, whose further evolution can cause chain scission, chain branching and/or crosslinking. These reactions produce changes in their thermal properties, depending on the material.
The melting (Tm) and crystallization (Tc) peak temperatures and crystallinity degree (X) of polyethylenes (HDPE, LDPE, LLDPE) irradiated with g-rays from a 60Cobalt source in air at a dose rate of 4.8kG/h decrease slightly when the absorbed dose increases (0-1000 kGy). These changes go from 122 -108 ºC, 105-93 ºC and 36-40 % in LLDPE, 130-124 ºC, 117-108 ºC and 66-53% in HDPE and 112-105 ºC, 96-87 ºC and 44-37 % in LDPE [22].
3.2 Irradiated Blends and Composites
Albano et al. [4] have reported the thermal behavior of composites of polypropylenes with sisal fiber (20 wt.%) and woodflour (40 wt.%), when irradiated at doses in the 0-70 kGy range, at a dose rate of 4.8 kGy/h, at room temperature in air. Values of melting peak temperatures of the composites under study are displayed in Table 14. As can be seen, these values remain constant at the lowest irradiation dose (10kGy) and then tend to decrease as the integral dose increases. This behavior is attributed to the oxidative degradation of PP when it is submitted to the irradiation dose, which promotes the scission of the polymer chains, which melt at lower temperatures.

In the thermograms obtained by Thermogravimetric Analysis (TGA) of PP filled with woodflour and sisal fiber, two decomposition stages can be identified (Figure 12). Due to this behavior, kinetic studies as well as a global analysis of the activation energy were carried out for every stage of the degradation process. Values of the activation energy of the first and the second decomposition stages, obtained through the McCallum-Tanners kinetic method [48], as well as the value of global Ea of the whole decomposition process of the PP/woodflour and PP/sisal fiber composites are presented in Tables 15 and 16.



The values of activation energy for the first stage correspond to the filler decomposition. These values exhibit a behavior fluctuating within the irradiation range of the samples. Depending upon the radiation dose, they range between 113 and 56 kJ/mol in the sisal fiber, and between 76 and 49 kJ/mol in the woodflour. However, such values seem to be independent of the radiation dose in composites with woodflour, but dependant on the dose in those composites with sisal fiber. Therefore, the value of Ea of sisal fiber is always higher than that of the woodflour; this implies that greater thermal stability has been achieved by the sisal fiber. The Ea values of the second decomposition stage correspond to PP. These values are higher than those obtained in the first stage and within the range obtained by Albano et al. [49]. A comparison of the values of Ea of PP in irradiated composites with woodflour or with sisal fiber shows that the activation energy in the former (PP/woodflour) ranges between 163-180 kJ/mol (Table 15), whereas in the latter is higher, i. e., between 180-214 kJ/mol (Table 16).
On the other hand, Albano et al. [8] analyzed the changes produced by irradiation in the Tm, Tc and X values of HDPE/hydroxyapatite composites obtained via solution in decalin. They also studied the thermal behavior of the HDPE used to prepare such composites, subjected to the same mixing (dissolution in decalin and precipitation, HDPEd) and irradiation conditions. Tables 17, 18 and 19 exhibit the thermal properties of the composites. As seen there, the Tm and Tc values remained unchanged after the radiation process. However, from Table 19 it is clear that the crystallinity degree showed a slight increase with filler addition that could be attributed to a minor nucleating effect of nanometric HA particles. When samples were exposed to gamma radiation, a decrease in crystallinity was observed in all the materials, probably due to the interruption of the more linear sequences of the polyethylene resulting from a chain scission process and/or crosslinking.



Figure 13 displays the DSC heating scans after applying the Successive Self-Nucleation and Annealing (SSA) thermal treatment fractionation to HDPE (previously dissolved and precipitated) irradiated at different absorbed doses. The development of this technique was reported by Müller et al. [50,51] and has been used to characterize semicrystalline polymers that are capable of undergoing molecular segregation during crystallization upon cooling from the melt [52,53].

At 50 and 100 kGy of absorbed doses, a broadening of the main melting peak and the appearance of a shoulder in the peak towards its lower temperature side are observed. It seems that the irradiation at 50 kGy, and even more at 100 kGy, could be producing an interruption and a slight decrease in the length of the crystallizing CH2 sequences, thus increasing the crystal populations that melt at lower temperatures (represented in that shoulder). Such a fact is consistent with the formation of some thinner lamellas at the expense of the disappearance of some of the thicker ones.
The thermal stability of the composites was determined through thermogravimetric analyses. In polymers, some factors such as the polymer/filler compatibility and their structures could affect the decomposition process. The initial decomposition temperatures (Tid) allow determining the beginning of the decomposition process of the composites (Table 20). Tid tends to increase slightly with HA additions as a consequence of a stabilizing effect provided by the filler. On the other hand, radiation tends to cause a slightly decrease in Tid, as a consequence of the degradation of the polymer.

Table 21 exhibits the activation energy of the composites at the different absorbed doses. The behavior is analogous to that of the initial decomposition temperatures. In non-irradiated samples, the thermal stability of the composites is improved with the inclusion of HA, reaching a plateau at 20 phr. In general, it can be said that HA is affecting the beginning of the degradation by retarding it. In general, all the composites irradiated at different absorbed doses are less stable and more prone to thermal degradation than their nonirradiated counterparts.

Other studies carried out by Ramírez et al. [11] found that neither Tm nor Tc of composites of polypropylene with hydroxyapatite show significant variations with the filler content. However, a slight decrease in Tm was observed with the increase in the integral dose in all the materials. This was attributed to polymer chain scissions produced by radiation, which decrease its molecular weight and generate defects. The crystallinity degree remained approximately constant with the filler content. This fact can be attributed to an inhomogeneous distribution of HA and to the formation of large agglomerates, which could possibly affect the formation of nucleation sites for crystallization. The radiation dose also showed very insignificant effects on the crystallinity degree, as it has also been reported in the literature, where only at very high radiation doses (>500kGy) important effects has been observed [30,44].
Table 22 shows the initial degradation temperature (Ti) and activation energy (Ea) values of the irradiated and non-irradiated samples. The values of Ti do not unfold significant changes up to 10 kGy of absorbed dose. Nonetheless, at 25 kGy, a more significant decrease of these values is observed for all materials. The activation energy values remain almost constant between 0 and 10 kGy for PP and PP-20%HA, indicating some thermal stability of these materials in this range of radiation. However, for higher radiation doses or higher filler contents, Ea decreases drastically. This result can be attributed to the effect of higher radiation doses on PP, and additionally, to an increased content of agglomerates for higher filler contents.

In composites of PMMA with seaweed residues, the addition of the filler does not modify the glass transition temperature of the polymer [9]. However, the radiation at 100kGy slightly decreased this parameter, from 102 to 98 °C, which confirms the degradation of the PMMA. Chipara [54] reported changes in Tg of polymers with irradiation: an increase when crosslinking is the predominant mechanism of degradation and a decrease when depolymerization or chain scissions are the prevailing mechanisms.
TGA analyses demonstrated that the initial decomposition temperature did not change significantly neither with the inclusion of the seaweed residues nor with the irradiation process. Nonetheless, when pure PMMA was irradiated at 100 kGy, a decrease in this parameter was reported due to its degradation (Table 23). Additionally, an increase in the activation energy with the amount of filler was found (Table 24), which demonstrates its stabilizing effect when added at concentrations of up to 10 phr. At higher amounts, a decrease in Ea is observed, which is attributed to possible filler-filler interactions and to an increased interfacial area that favors the degradation of the composite. As expected, Gamma radiation produced a decrease in the thermal stability of the PMMA and its composites, which is clearly revealed by the significant drop of the Ea of the irradiated pure PMMA and a gradual drop of the composites with the increase in the integral dose.


Regarding the thermal behavior of blends of PS/PP, Albano et al. [15] observed that the glass transition temperature of pure PS remained approximately constant when irradiated in the absorbed dose interval of 0-70 kGy; increased from 100 °C up to 105 °C within the dose range between 70 and 400 kGy, and then stayed constant at that value at 1300 kGy. This is indicative of a crosslinking process in this irradiation dose range, which is well documented [55] and demonstrated through the yellowish coloration of the samples. On the other hand, the melting peak temperature of irradiated neat PP (figure 14) showed a continuous decrease, varying from 159 °C at 0 kGy to 152 °C at 70 kGy, and to 139 °C at 1300 kGy. Similar behavior was observed in PP in the blends under study (figure 15). These results confirm that within the range of high irradiation doses, PP shows a highly oxidative degradation (chain scission). In addition, the Tg values of PS in the blends show very slight increases, the variation being 3 °C.


On the other hand, the crystallinity degree of PP in the blends (Table 25) was found to decrease as a function of the irradiation dose, which is related to chain scission reactions. Irradiation decreases the rate of transport of crystallizable segments of PP to the crystallization front during the cooling process of these blends in the DSC. Changes in crystallinity of pure PP are not observed, although this polymer shows a degradative process of chain scissions which increases with the irradiation dose. According to Dole [19], doses higher than 1000 kGy are required to detect variations in PP crystallinity degree.

The scission and crosslinking processes produced in PP, PS and their blends, are due to: 1) tertiary polystyryl radicals, which are found initially by gamma irradiation because of scission of the C-H bond, 2) the ciclohexadienyl radicals that have also been found and are attributed to the addition of a hydrogen radical to the phenyl ring. Close to room temperature and in the absence of oxygen, polystyryl radicals react by crosslinking, which should happen inside the specimens due to the low oxygen concentration and its slow diffusion inside the sample; and 3) the alkyl radical in PP, which transforms into a peroxy radical [56]. According to Gugumus [57], PP hydroperoxides, in the solid phase, can interact with two adjacent atoms of the polymer chain and the result would be chain scission and the formation of carbonyl groups, thus resulting in a bimolecular kinetic mechanism involving intramolecular and intermolecular hydroperoxide decomposition reactions.
Since blends of different polyethylenes are widely used, Quero et al. [17] analyzed the peak melting temperatures of irradiated LDPE/HDPE samples (Table 26). Prior to the irradiation process, the blends were prepared with different thermal histories, by fast cooling and slow cooling from the melt. From their results, it is interesting to observe that in the case of non-irradiated LDPE, the rapidly cooled sample displays a slightly higher melting peak temperature. Higher temperature peaks for non-irradiated quenched branched polyethylene samples than those found for slowly cooled samples, as shown in Table 26, have been reported in the past and explained on the basis of crystalline structure reorganization during heating [58]. On the other hand, the non-irradiated and irradiated samples of the 50/50 LDPE/HDPE blend showed contrasting behaviors. In non-irradiated samples, the peak melting temperature was located at higher temperatures for the quenched samples, whereas for irradiated ones, the peak melting temperature of the slowly cooled was higher or at about the same temperature than that of the quenched sample. For neat HDPE, changes in peak melting temperatures are more pronounced for the irradiated samples. Therefore, the results presented in Table 26 indicate that changes in the PEs chemical structure do occur due to Gamma irradiation. Such changes may be the formation of enough crosslinks to prevent crystals from reorganization or to inhibit crystalline rearrangement during heating in the DSC. HDPE, being a more crystalline material, however, exhibits a different behavior. As expected, the peak melting temperature of the rapidly cooled HDPE sample is lower than that of the slowly cooled. The crystals, due to their higher perfection, are less prone to rearrangement during heating. Irradiation has the effect of increasing the melting temperature for the slowly cooled HDPE sample, whereas the rapidly cooled one follows the trend exhibited by the branched samples. For HDPE, with a higher crystallinity degree, it would be expected that the heat resistance of large and perfect crystals surrounded by a crosslinked amorphous layer is increased.

Regarding the effect of HDPE content in the blend (0-100 %), the authors report that the initial crystallinity degree varied between 43 and 64 % for rapidly cooled samples, whereas for the slowly cooled blends it varied between 44 and 71 %. Measurements of the crystallinity degree failed to show significant changes with irradiation (less than 10%). These results are in agreement with those obtained previously which showed that changes in the crystallinity degree occurred at higher doses than those studied in this work [59]. It turned out to be more dependent on the type of cooling applied to the blends: larger values were found for slowly cooled samples.
Perera et al. [60] reported the thermal behavior of irradiated PP/SBS blends. They indicate that the shape of the calorimetric melting curve provides valuable information about the thermal history and structural characteristics of the sample. Figure 16 displays the melting endotherms of pure PP irradiated at 25, 50 and 100kGy. Those endotherms show well-defined peaks whose melting peak temperatures and enthalpy (DHm) values are reported in Table 27. The shown values of DHm are similar for the irradiated and unirradiated PP samples, indicating that the crystallinity content is unaffected by the radiation, no matter what the radiation dose is (25, 50 or 100 kGy). In the same figure 16, it is also shown how the non irradiated PP presents just one melting peak, whereas the irradiated PP at the different doses used reflects two melting peaks, which are displaced towards lower temperature values as the irradiation doses increase. Duplication or multiplication of melting peaks can be attributed to structural reasons. Multiple endotherms are observed in a wide variety of semicrystalline polymers and can arise from segregation effects by molecular weight, among other parameters.


The fact that two peaks whose resolutions increase with the radiation dose are present in melting thermograms suggests that two different and well defined crystalline populations with differing lamellar thickness coexist, one thicker with more perfect crystals melting at higher temperatures, and the other one thinner, with less perfect crystals melting at lower temperatures. This difference in crystalline populations can then be attributed to the decrease in the molecular weight of the PP, due to the chain scission brought about by the radiation process [61,62], which in turn lowers the onset temperatures displayed in Table 27 for both populations.
Regarding the melting thermograms of the blends containing 20 phr of SBS (figure 17), a decrease in the melting temperature can be seen as the radiation dose is increased (see Table 28). Furthermore, a shoulder appears which evolves as a peak at 100 kGy. Similar melting thermograms were recorded in the blends containing 30 and 40 phr of SBS (Tables 29 and 30). In these last two cases, the addition of 30 and 40 phr of SBS to PP results in a slight decrease in the crystallinity degree of PP in the nonirradiated samples.




On the other hand, a small decrease in the melting enthalpy of PP with the radiation process resulted in the blend containing 20 phr of SBS, which indicates a decrease in its crystallinity degree. When the melting enthalpy of this blend and that of pure PP, both irradiated, are compared, it can be confirmed that the SBS copolymer appears to be modifying slightly the effect of the radiation onto the PP. Saroop and Mathur [63] found that a crosslinked structure produced under dynamic vulcanization in the same copolymer in PP/SBS blends may restrict the spherulitic growth and hence, decreases the crystallinity degree of PP. Same results seems to be obtained in this work when ionizing radiation was employed.
From Tables 27, 28, 29 and 30, it can be concluded that blending PP with SBS produces a decrease in its crystallinity degree when irradiated and that changes in those crystallinity degrees depend on the SBS content and the integral doses used. Silva et al.[64], demonstrated that the free radical concentration in PP/SBS blends increased with the dose, as expected. They also found a slight decrease in the total free radical concentration when the amount of SBS was increased, and that pure PP had a higher free radical concentration than its blends with SBS. This fact was attributed to the crystalline character of PP, whereas SBS is amorphous. On the other hand, the benzene ring present in SBS, due to stability and steric reasons, hinders the oxygen diffusion and its reaction in the irradiated materials, thus decreasing the amount of peroxy radicals formed when SBS is present. Additionally, the SBS copolymer acts as a diluent in the PP crystallization process, increasing its amorphous character, which brings about the fact that if the radicals are indeed formed, they will recombine quickly, forming stable structures.
Through the analysis of the results obtained using the SSA fractionation technique proposed by Muller et al. [50], it is easy to ascertain the heterogeneity in crystalline populations as a consequence of chemical modifications in the polyolefins, either after grafting and/or peroxide treatment or after secondary or collateral reactions. The fractionated polymers show many separated melting peaks, corresponding to the melting of crystals of different lamellar thickness, which are limited by chain branches or functional groups that are excluded from the crystalline structure.
Sánchez et al. [18] applied the SSA technique to LDPE grafted with diethyl maleate using Gamma rays (figure 18). They observed that the heating scans obtained after applying the SSA treatments revealed the presence of nine melting endotherms corresponding to chain segregation as a function of chain imperfections and branching. Grafted LDPE displayed a significant decrease in the relative height of the endotherm with higher melting peak temperature, and an increase in the height of the endotherms with lower melting temperatures. This effect is more noticeable as the radiation dose increases and is attributed both to higher grafting degrees and to chemical modifications such as branching/crosslinking in the main chain, which proceed through secondary carbons [65].

Quantitatively, this behavior is evidenced in the peak areas of each endotherm (Table 31), where those of the higher melting temperature peaks decrease as new and less perfect crystalline populations are created. The largest area decrease of the higher temperature peak was obtained in the sample of LDPE with the higher grafting degree at 400 kGy. The melting peak temperatures of all peaks remained almost unchanged in all samples, because a change would only be a consequence of the employed self-seeding temperature.

3.3 Conclusions
From the obtained results, it can be inferred that depending on the polymer-filler interaction, it is possible to have different behaviors in the activation energy (Ea) with the irradiation dose. In those composites of PP with woodflour and sisal fiber, even though both filler are organic in nature, the behavior is different. Probably, the L/D ratio of the filler particles is determining it. On the other hand, in composites of HDPE/HA obtained via solution, and in those of PP/HA and PMMA with seaweed residues irradiated at different doses, a decrease in the Ea values is seen, which indicates that in these cases the effect of the radiation is significant. In other words, the degradation process speeds up, and its rate depends on the preparation technique of the composites, as well as on the chemical structure of the polymer matrix.
Regarding the thermal behavior, in general, a displacement of the endotherms towards lower temperatures and a decrease in the crystallinity degree of blends of PS/PP and PP/SBS was observed as a consequence of the radiation process. In the case of LDPE grafted with diethyl maleate using gamma rays, modifications of the main chain were produced as the radiation dose was increased. These chain modifications bring about changes in the endotherms obtained through the SSA technique.
4.-REFERENCES
1. Spadaro G, Valenza A. Polym. Deg. Stab. 2000; 67: 449-454. [ Links ]
2. Chang Z, Laverne J. J. Polym. Sci. A: Polym. Chem. 2000; 38: 1656-1661. [ Links ]
3. Zhang XC, Buthler MF, Cameron RE. Polym. Int. 1999; 48: 1173-1178. [ Links ]
4. Albano C, Reyes J, Ichazo M, González J, Brito M, Moronta D. Polym. Deg. Stab. 2002; 76: 191-203. [ Links ]
5. Cataño L, Albano C, Karam A, Dominguez N. Mechanical Behavior of Irradiated Polyethylenes. In: Proceedings of PACIFICHEM 2005. Honolulu (EE.UU.) 2005, Program Number: 720. [ Links ]
6. Albano C, Perera R, Karam A, Sánchez Y, Silva P. Nuc. Instr. and Meth. B. 2007; 265: 265-270. [ Links ]
7. Albano C, Reyes J, González J, Ichazo M, Poleo R, Davidson E. Polym. Deg. Stab. 2001; 73: 39-45. [ Links ]
8. Albano C, Karam A, Perera R, Gonzalez G, Dominguez N, González J, Sánchez Y. Nuc. Instr. and Meth. B. 2006; 247: 331-341. [ Links ]
9. Albano C, Karam A, Domínguez N, Sánchez Y, González J, Revista de la Facultad de Ingeniería UCV. 2006; 21 (1): 21-28. [ Links ]
10. Cataño L, Albano C, Karam A, Domínguez N, Sánchez Y, González J. Nuc. Instr. and Meth. B. 2005; 236: 348-353. [ Links ]
11. Ramírez C, Albano C, Karam A, Domínguez N, Sánchez Y, González J. Nuc. Instr. and Meth. B. 2005; 236: 531-535. [ Links ]
12. González J, Albano C, Candal MV, Ichazo MN, Hernández M. Nuc. Instr. and Meth. B. 2005; 236: 354-358. [ Links ]
13. Albano C, Reyes J, Ichazo MN, González J, Rodríguez M. Nuc. Instr. and Meth. B. 2003; 208: 485-488. [ Links ]
14. Albano C, Reyes J, Ichazo M, González J, Hernández M, Rodríguez M. Polym. Deg. Stab. 2003; 80: 251-261. [ Links ]
15. Albano C, Reyes J, Ichazo M, González J, Hernández M. Revista de la Facultad de Ingeniería UCV. 2005; 20 (2): 85-94. [ Links ]
16. Albano C, Perera R, Silva P, Sánchez Y. Polym. Bull. 2006; 57: 901-912. [ Links ]
17. Quero E, Puig CC, Albano C, Karam A. Polym. Bull. 2007; 59: 517-526. [ Links ]
18. Sánchez Y, Albano C, Perera R, Karam A, Silva P. Macromol. Symp. 2007; 257: 139-146. [ Links ]
19. Dole M. The Radiation Chemistry of Macromolecules, USA: Academic Press, 1972, Vol: I. [ Links ]
20. Dole M, The Radiation Chemistry of Macromolecules, USA: Academic Press, 1973, Vol: II. [ Links ]
21. Ivanov VS, Radiation-chemical transformation of polymers. In: Hi of Jonge CR (ed.), New Concepts in Polymer Science. VSP, Utrecht (1992), chapter 3. [ Links ]
22. Albano C, Perera R, Silva P, Sánchez Y. Polym. Bull. 2003; 51: 135-142. [ Links ]
23. Kusumoto N, Yamamoto T, Takayanagi M. J. Polym. Sci. A: Polym. Chem. 1971; 2: 1173-1190. [ Links ]
24. Dalvag H, Klason C, Stromvall HE. Int. J. Polym Mater. 1985; 11: 9-38. [ Links ]
25. Raj RG, Kokta BV, Danault C. Int. J. Polym Mater. 1989; 12: 239-250. [ Links ]
26. Bisanda ETN, Ansell MP. Compos. Sci. Technol. 1991; 41: 165-168. [ Links ]
27. Slovokhatova NA, Il´icheva ZF, Valisiev LA, Kargin VA. Polym. Sci. USSR, 1964; 6: 671-678. [ Links ]
28. Zamotaev P, Chodak I, Mityukhin O, Chorvath I. J. Appl. Polym. Sci. 1995; 56: 935-946. [ Links ]
29. Smirnov LP, Deyun EV. Polym. Sci. Serv. A. 1999; 41 (5): 782-788. [ Links ]
30. Chapiro A. Radiation Chemistry of Polymeric Systems. New York (USA): John Wiley &Sons, Interscience, 1962, chapters VIII and IX. [ Links ]
31. Geymer DO. Makromol. Chem. 1967; 100: 186-188. [ Links ]
32. Miller AA, Lauton EJ, Balwit JS. J. Polym. Sci. 1954; 14: 503-504. [ Links ]
33. Guan RJ. J. Appl Polym. Sci. 2000; 76: 75-82. [ Links ]
34. Chipara MD, Grecu VV, Chipara MI, Ponta C, Reyes Romero J. Nuc. Instr. and Meth. B. 1999; 151: 444-498. [ Links ]
35. Arroyo M, Avah F, Rev. Plast. Modern. 1988; 383: 705-714. [ Links ]
36. Tanner KE, Downes R, Bonfield W. Br. Ceram. Trans. 1994; 93(3): 104-107. [ Links ]
37. Smallwood HM. J. Appl. Phys. 1944; 15: 758-766. [ Links ]
38. Guth E. J. Appl. Phys. 1945; 16: 20-25. [ Links ]
39. Nicolais L, Narkis M. Polym. Eng. Sci. 1971; 11: 194-199. [ Links ]
40. Maiti SN. Lopez BH. J. Appl. Polym. Sci. 1992; 44: 353-356. [ Links ]
41. Kunori T, Geil PH. Macromol. Sci. Phys. B. 1980; 18: 135-175. [ Links ]
42. Nielsen L. Mechanical Properties of Polymer and Composites, New York (USA): Marcel Dekker, 1974, chapter 7. [ Links ]
43. Qing L., Winjn J., Clemens A., J. Biomater. Mater. Res. 1997; 40: 524-530. [ Links ]
44. Miguez J, Biasotto E, Abrahão R. Polym. Deg. Stab. 2002; 69: 217-222. [ Links ]
45. Hlavata D, Horak Z, Folt V. Polym. Networks Blends. 1995; 37(1): 15-19. [ Links ]
46. Vishwa Prasad A, Singh RP. J. Appl. Polym. Sci. 1998; 70: 637-645. [ Links ]
47. Schnabel W, Levchik G, Wilkie C, Jiang D, Levchik S. Polym. Deg. Stab. 1999; 63: 365-375. [ Links ]
48. McCallum JP, Tanner J. Eur. Polym. J. 1970; 6: 1033-1039. [ Links ]
49. Albano C, González J, Ichazo M, Kaiser D. Polym. Deg. Stab. 1999; 66: 179-190. [ Links ]
50. Müller AJ, Hernández ZH, Arnal ML, Sánchez JJ. Polym. Bull. 1997; 39: 465-472. [ Links ]
51. Müller AJ, Arnal ML. Prog Polym. Sci. 2005; 30: 559-603. [ Links ]
52. Arnal ML, Hernández ZH, Matos M., Sánchez JJ, Méndez G, Sánchez A, Müller AJ. Proceedings of the 56th Annual SPE Technical Conference (ANTEC) 1998. Georgia (USA): 1998, p. 2007-2011. [ Links ]
53. Villarreal N, Pastor JM, Perera R, Rosales C, Merino JC. Proceedings of the 60th Annual SPE Technical Conference (ANTEC) 2002. San Francisco (USA): 2002, p. 3812-3816. [ Links ]
54. Chipara MI, Physica B: Condensed Matter. 1997; 263: 234-236. [ Links ]
55. Grassie N, Weir NA. J. Appl. Polym. Sci. 1965; 9: 999-1003. [ Links ]
56. Bikales M, Mengles O. Encyclopedia of Polymer Science and Engineering, 2nd ed. (Canada): Wiley-Interscience Publication, 1989, Vol 16. [ Links ]
57. Gugumus F. Polym. Deg. Stab. 2001; 74: 327-339. [ Links ]
58. Peeters M, Goderis B, Vonk C, Reynaers H, Mathot M. J Polym. Sci. B: Polym. Phys. 1997; 35: 2689-2713. [ Links ]
59. Vaughan AS, Ungar G, Bassett DC, Keller A. Polymer. 1985; 26:726-732. [ Links ]
60. Perera R, Albano C, Gonzalez J, Silva P, Ichazo M. Polym. Deg. Stab. 2004; 85: 741-750. [ Links ]
61. Varga J, Crystallization, melting and supermolecular structure of isotatic polypropylene In: Karger-Kocsis J. (ed.), Polypropylene: Structure, Blends and Composites. London (UK): Chapman & Hall, 1995, Vol. I: Structure and Morphology, p. 56-115. [ Links ]
62. Phillips RA, Wolkowicz MD, Structure and Morphology In: Moore EP. (ed.), Polypropylene Handbook. New York (USA): Hanser, 1996, p. 113-176. [ Links ]
63. Saroop M, Mathur GN. J. Appl. Polym. Sci. 1999; 71 (2): 151-161. [ Links ]
64. Silva P, Albano C, perera R,González J, Ichazo M. Nuc. Instr. and Meth. B. 2004; 226; 320-326. [ Links ]
65. Márquez L, Rivero I, Müller AJ. Macrom. Chem. Phys. 1999; 200: 330-337. [ Links ]
