










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Latinoamericana de Metalurgia y Materiales
versión impresa ISSN 0255-6952
Rev. LatinAm. Metal. Mater. v.30 n.2 Caracas dic. 2010
Caracterización de catalizadores 0,8% Pd/TiO2 sol-gel usados en reacciones de hidrodecloración catalítica en fase líquida
Alba N. Ardila Arias
Grupo de Investigación en Química Básica y Aplicada a Procesos Bioquímicos, Biotecnológicos y Ambientales, Facultad de Ciencias Básicas Sociales y Humanas, Politécnico Colombiano Jaime Isaza Cadavid, Medellín, Colombia
E-mail: anardila@elpoli.edu.co, albanellya@gmail.com
Publicado On-Line el 30-Jul-2010
Disponible en: www.rlmm.org
Resumen
Se caracterizó por una amplia gama de técnicas fisicoquímicas muestras seleccionadas del catalizador 0.8% Pd/TiO2 sol-gel fresco y usado en reacciones de hidrodecloración catalítica de tetracloroetileno en fase líquida con hidrogeno puro en presencia de NaOH bajo condiciones de reacción moderadas. El objetivo del trabajo fue conocer las propiedades fisicoquímicas del catalizador que le permitieron tener un alto desempeño catalítico e identificar los efectos producidos por las condiciones de la reacción sobre dichas propiedades. Los resultados indican que el método de síntesis y de pretratamiento utilizados para preparar el catalizador, permitieron obtener un material mesoporoso, con un área superficial relativamente alta, débilmente hidroxilado con acidez tipo Lewis, con una fase activa moderadamente dispersa y con especies de Pd0 y Pd2+ en la superficie. No obstante, las caracterizaciones realizadas al catalizador usado revelan ciertos cambios como: disminución en el área específica, aumento de la acidez superficial, presencia de algunos compuestos orgánicos, disminución de la dispersión y con ello el aumento en el tamaño de las partículas de paladio. Sin embargo, algunas características como la estructura cristalina del soporte, el contenido metálico, la forma de las partículas de paladio y sus estados de oxidación permanecieron casi constantes.
Palabras Claves: técnicas fisicoquímicas, catalizador 0.8% Pd/TiO2, sol-gel, hidrodecloración, tetracloroetieno.
Abstract
Selected samples of fresh and used 0.8% Pd/TiO2 sol-gel catalyst were characterized by a wide range of physicochemical techniques. This catalyst was used in reactions of hydrodechlorination of tetrachloroethylene in the liquid phase, with pure hydrogen, in the presence of NaOH, under mild conditions. The purpose was to study the physical and chemical properties of the catalyst which enabled a high catalytic activity and identify the effects of the reaction conditions on these properties. The results indicate that the method of synthesis and pre-treatment used to prepare the catalyst helped to obtain a mesoporous material, with a relatively high surface area, weakly hydroxylated with type Lewis acidity and with Pd0 and Pd2+ superficial species. However, the characterizations of the used catalyst showed some changes, such as: decrease in surface area, increase in surface acidity, the presence of some organic compounds, decrease in dispersion and, thereby, increase in the size of palladium particles. However, some features like the crystalline structure of the support, the metal charge, the shape of palladium particles and their oxidation states remained almost constant.
Keywords: physicochemical techniques, 0.8% Pd/TiO2 sol-gel catalyst, hydrodecholorination, tetrachloroethylene.
Recibido: 15-Ene-2009; Revisado: 02-Dic-2009; Aceptado: 25-Ene-2010
1. INTRODUCCIÓN
Los compuestos organoclorados ligeros constituyen uno de los tipos de contaminantes atmosféricos y acuíferos que más preocupación han despertado en las últimas décadas, pues incluyen una gran cantidad de productos tóxicos para el ser humano, cuya velocidad de degradación en suelos y aguas es baja, lo que agrava sus efectos. Estos compuestos una vez en la atmósfera, contribuyen a la degradación de la capa de ozono estratosférico, al calentamiento global del planeta y a la formación del smog fotoquímico, además de efectos directos sobre la salud de los seres vivos cuando están presentes en el agua [1-3].
Dada la toxicidad de estas sustancias, el método de tratamiento más adecuado es la destrucción química. Con la hidrodecloración catalítica en fase líquida, se han obtenido muy buenos resultados, lográndose degradar completamente diferentes tipos de estos contaminantes. Este método consiste en hacer reaccionar el compuesto organoclorado con hidrógeno, produciéndose hidrocarburos ligeros (aprovechables por combustión o como materia prima) y cloruro de hidrógeno, fácilmente eliminable por absorción con álcalis. No obstante, para que esta técnica sea aplicable, se debe disponer de un catalizador muy activo y resistente al envenenamiento por HCl, además de diseñar el equipo para que el aprovechamiento de hidrógeno sea máximo, evitando así el encarecimiento del proceso [4,5].
Entre los catalizadores estudiados en las reacciones de hidrodecloración catalítica en fase líquida, los más usados son los de hidrotratamiento, de níquel y de metales preciosos como platino, rutenio, rodio y paladio soportados sobre alúmina, titania, sílice y carbón activado [6-9]. No obstante, algunas investigaciones realizadas han demostrado que los catalizadores de paladio soportado sobre alúmina y titania son los más activos y selectivos en este tipo de reacciones, sin embargo, en las mismas investigaciones se ha reportado que después de cierto tiempo de reacción los catalizadores se desactivan [10-14]. Se ha especulado que la actividad y desactivación de los catalizadores de paladio durante el curso de las reacciones de hidrodecloración catalítica en fase líquida se puede deber a varios factores como: número y clase de especies de paladio presentes, variables de preparación del catalizador (sal precursora, tratamientos térmicos y reductivos) tipo y naturaleza del soporte, carga y dispersión del metal, temperatura de la reacción, solvente y donor de hidrógeno utilizados y presencia de ciertas sustancias en el medio de reacción como el HCl producido, la base adicionada o las sales formadas por la adición de dichos álcalis, entre otros [15-21]. No obstante, para comprender realmente el comportamiento catalítico de estos materiales en el ambiente de la reacción, se hace necesario conocer sus características fisicoquímicas antes y después de usarlos en la misma, sólo así se podría establecer relaciones entre sus propiedades fisicoquímicas y su desempeño catalítico, así como también, esclarecer las causas de su desactivación, proporcionando de esta manera la clave para la predicción de las propiedades catalíticas de un catalizador óptimo para las reacciones de hidrodecloración catalítica en fase líquida.
En el presente estudio, se reportan los resultados obtenidos en las caracterizaciones realizadas a muestras seleccionadas del catalizador 0,8% Pd/TiO2 sol-gel fresco y usado. Estos materiales se ensayaron en reacciones de hidrodecloración catalítica de tetracloroetileno (71.5 mM) en fase líquida en metanol y una concentración inicial de NaOH igual a 12.06 mM con hidrógeno puro, bajo condiciones moderadas de reacción, obteniéndose un 94% de conversión del compuesto organoclorado en tan solo 10 minutos. Es importante, resaltar que con este catalizador, el tatracloroetileno fue degradado completamente a etileno, sin la formación de algún producto organoclorado intermediario. Los resultados y detalles de este estudio se encuentran publicaos en [1,22]. El objetivo del presente estudio es conocer las propiedades fisicoquímicas de los catalizadores que le permitieron tener un alto desempeño catalítico e identificar los efectos producidos por las condiciones de la reacción sobre dichas propiedades.
2. PARTE EXPERIMENTAL
2.1 Síntesis del soporte TiO2 sol-gel
El soporte de titania se sintetizó por el método solgel, según el procedimiento reportado en [1,22], para lo cual se partió de una suspensión de 22.9 mL de butanol y 9.12 mL de butóxido de titanio, y se ajustó el valor del pH de la síntesis a 9.0 adicionando hidróxido de amonio, posteriormente para iniciar la etapa de hidrólisis y condensación se adicionó 11.5 mL de agua. En el proceso sol-gel, la temperatura de calcinación es una variable potencialmente importante a la hora de remover las moléculas orgánicas del producto final y completar la cristalización, ya que una baja temperatura de calcinación (> 600°C), podría no ser suficiente para transformar todos los titanatos amorfos en fase cristalinas [1,22]. Por lo tanto, el soporte sol-gel obtenido se calcinó en aire estático hasta 600ºC con una velocidad de calentamiento de 10ºC/min.
2.2 Preparación del catalizador 0,8% Pd/TiO2 sol-gel
Para la preparación del catalizador 0.8% Pd/TiO2 sol-gel, se utilizó el método de impregnación húmeda incipiente del soporte previamente sintetizado, usando una solución de acetil acetonato de paladio en acetona [1,22]. Posteriormente, el catalizador sintetizado se calcinó a 300°C en aire y luego se redujo en flujo de H2/N2 al 5% hasta 300°C durante una hora a 2°C/min, con el fin de obtener paladio metálico.
2.3 Caracterización del catalizador 0.8% Pd/TiO2 sol-gel
Muestras seleccionadas de catalizadores frescos y usados se caracterizaron por una amplia gama de técnicas fisicoquímicas como titulación de masas, espectroscopía de absorción atómica (AAS), quimisorción de H2, adsorción-desorción de N2, difracción de rayos X (DRX), reducción con hidrógeno a temperatura programada (TPR-H2), análisis termogravimétrico (TGA-DTG), espectroscopía de reflectancia difusa UV-Vis (UVVis DRS), desorción de NH3 a temperatura programada (TPD-NH3), espectroscopía infrarroja de NH3 y CO adsorbidos y microscopía de transmisión electrónica (TEM).
El método de titulación de masas se empleó para determinar el punto isoeléctrico del soporte y los catalizadores. El análisis consistió en la medición del pH resultante del equilibrio de diferentes cantidades de masas de la misma muestra (20-240 mg) en un volumen dado de agua destilada que se agitó por espacio de 24 h. El pH de cada suspensión acuosa se midió con un pH-metro Metrohm modelo 691. Los difractogramas de las muestras se obtuvieron en un difractómetro Siemens modelo D- 500, empleando una fuente de radiación Ka del Cu con filtro de Ni a 40kV y 40 mA y una longitud de onda igual a 1.5406 Å, con una velocidad de barrido de 2 grados por minuto. Las medidas de área específica y porosidad de los catalizadores se llevaron a cabo con N2 a 77K en un equipo Quantachrome Autosorb Automated. La carga de paladio se determinó con un espectrofotómetro de absorción atómica Varian, modelo SpectraAA- 20SF. Los análisis de quimisorción con hidrógeno y desorción de NH3 con temperatura programada (TPD) se llevaron a cabo en un equipo Micromeritics AutoChem II 2920, para esto primero se realizó un pretratamiento de los sólidos en helio a 10°C/min hasta 300°C y se dejó 30 minutos a esta temperatura, con el fin de remover las impurezas y humedad presentes en el catalizador. Posteriormente, se disminuyó la temperatura hasta 25°C con el mismo flujo de helio. Luego se puso en contacto con 50mL/min de 0,3% NH3/He por 90 minutos, para asegurar la saturación completa de los catalizadores. Luego las muestras se purgaron con helio durante 60 minutos para remover el amoníaco débilmente adsorbido. Por ultimo, se realizó la desorción del amoníaco aumentando linealmente la temperatura del sistema (10°C/min) hasta 900°C. La reducción con hidrógeno a temperatura programada (TPR) se realizó con 10% H2/Ar con un flujo de 25 mL/min con un programa de temperatura ascendente a una rampa de 10ºC/min desde temperatura ambiente hasta 900°C y un detector de conductividad térmica (TCD). Las medidas de reflectancia difusa se realizaron directamente sobre las muestras en polvo a temperatura ambiente en un espectrofotómetro Cary/5E de Varian. Los análisis termogravimétricos se realizaron en un equipo 2950 TGA HR V6.1ª, modulado hasta 1000ºC. Los análisis IR de CO y NH3 adsorbidos, se realizaron en un equipo Bruker, IFS/ 166, las muestras se prepararon en forma de pastillas auto-soportadas y antes de realizar la medición, las muestras se calentaron a 500ºC en flujo de aire seco por una hora en una celda IR, esto con el fin de eliminar la mayor cantidad de agua posible, seguidamente se enfrió la muestra en flujo de nitrógeno hasta temperatura ambiente, finalmente se expuso la muestra con NH3 o CO por 1 hora para tomar la evolución de la señal con el tiempo, los espectros se grabaron en un intervalo entre 400 y 4000 cm-1. Por último, los catalizadores en polvo se observaron en un microscopio electrónico de transmisión Carl Zeiss, modelo EM910.
3. RESULTADOS Y DISCUSIÓN
3.1 Titulación en masas
La Figura 1 muestra las curvas del pH de equilibrio de la suspensión acuosa versus la cantidad de cada muestra de catalizador adicionada a la mezcla. Las mesetas de las curvas de titulación de masas corresponden al punto isoeléctrico de las muestras; los valores son 6.3, 6.0 y 5.7 para el soporte, el catalizador fresco y usado, respectivamente. El valor del punto isoeléctrico de los catalizadores analizados, evidencia que los materiales son de naturaleza anfotérica, característica que les confiere una alta actividad en los procesos de hidrodecloración en fase líquida, ya que pueden adsorber tanto especies catiónicas como aniónicas presentes en la mezcla de reacción [23]. Se ha discutido que la acidez del soporte tiene gran influencia en la resistencia que se pueda alcanzar hacia productos que se generan en las reacciones de hidrodecloración catalítica, como es el caso del cloruro de hidrógeno y el coque. En soportes de naturaleza muy ácida se ha visto que se pueden formar altas cantidades de depósitos carbonosos, mientras que los soportes de naturaleza básica y anfotérica tienen mayor resistencia al tolerar mejor la presencia del HCl, aumentando así la actividad del sistema [24,25,26]. Según Aramendía et al. [27], las propiedades ácido-base del soporte tienen una gran influencia en las reacciones de hidrodecloración. Dichos autores al comparar diferentes catalizadores con diferentes propiedades ácido-base, encontraron que las más altas conversiones de clorobenceno fueron obtenidas con los soportes de carácter anfotérico.
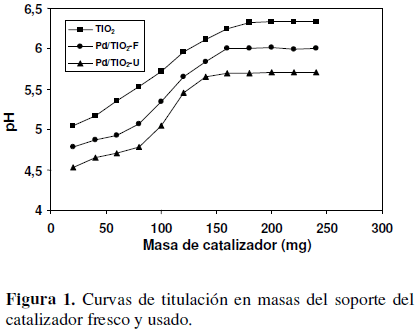
El punto isoeléctrico experimentalmente determinado para la titania, es consistente con los datos reportados en la literatura, los cuales indican que el punto isoeléctrico de la TiO2 sol-gel varía en un intervalo de pH entre 5.0 y 6.8 [18,28]. La suave disminución en el valor del punto isoeléctrico del catalizador fresco con respecto al soporte, sugiere que la impregnación del soporte con el metal produjo un aumento suave en la acidez del catalizador. Igualmente, el valor del punto isoeléctrico del catalizador usado es menor que el del fresco, indicando que durante la hidrodecloración catalítica aumentó la acidez del catalizador, lo cual se atribuye a la adsorción por parte del catalizador, de los iones cloruro producidos durante la reacción (Reacción 1), lo que a su vez provoca una disminución en el punto isoeléctrico del sólido [24,28].
C2Cl4 + 5H2 + 4NaOH Û C2H6 + 4NaCl + 4H2O
(Reacción 1)
3.2 Difracción de rayos X (DRX)
La titania naturalmente presenta tres estructuras cristalinas: anatasa, brookita y rutilo, no obstante, varias investigaciones han demostrado que la titania anatasa es la fase que muestra mejor desempeño catalítico en las reacciones de hidrodecloración catalítica, por lo cual es la fase deseada [19,29]. Sin embargo, la obtención de este material depende de varios factores, especialmente, del método de síntesis y del tratamiento térmico aplicado. El método sol-gel, se ha vislumbrado como un procedimiento adecuado para obtener titania anatasa con alta área específica, estabilidad térmica y química y propiedades ácido-base, las cuales son imprescindibles para su buen desempeño catalítico [30,31].
Aunque los patrones DRX del soporte y de los catalizadores fresco y usado mostraron algunas líneas de difracción de TiO2-rutilo, los picos con mayor intensidad en 2q = 25, 38, 48, 54 y 63°, son característicos de la TiO2-anatasa (Figura 2). La cantidad de anatasa presente en los catalizadores se calculó con la Ecuación 1 y representa un 89.4% con respecto a todo el catalizador, siendo el porcentaje restante correspondiente a la fase rutilo.


Los resultados obtenidos son consistentes con el tratamiento térmico aplicado a las muestras (600ºC), ya que según se ha reportado [31,32], la temperatura a la cual empieza a ocurrir el cambio de titania anatasa a titania rutilo es 700ºC. Esto indica que el procedimiento de síntesis y tratamiento de los catalizadores fue adecuado para la obtención de TiO2-anatasa; fase deseada por sus aplicaciones catalíticas, especialmente por su alta estabilidad y área específica. Al comparar el patrón de difracción del catalizador fresco con el del usado, los resultados obtenidos revelan que no hubo cambios cristalográficos en el soporte (TiO2) durante la reacción de hidrodecloración.
3.3 Adsorción-desorción de nitrógeno
La Figura 3 presenta los resultados de las isotermas de adsorción-desorción de nitrógeno a 77 K para el soporte y los catalizadores fresco y usado. Las isotermas de adsorción de nitrógeno por gramo de catalizador exhibieron un rápido aumento del volumen adsorbido a presiones relativas cercanas a 1, las muestras exhiben un comportamiento que se asemeja a una isoterma tipo IV, de acuerdo a la clasificación de la IUPAC, característica de sólidos mesoporosos [10,26,32]. Además, se observa adsorción en multicapas hasta que inicia la condensación capilar. Sin embargo, al comparar la forma de las isotermas del soporte con las del catalizador impregnado, se encuentra cierta divergencia, lo que nos lleva a suponer que la incorporación del paladio modificó suavemente las propiedades textuales de la titania.

En la misma Figura se puede observar la forma de la histéresis, en la cual que ambas curvas son bastantes verticales y casi paralelas, lo que corresponde a la histéresis tipo H1, de acuerdo con la clasificación IUPAC, que es característica de sólidos con poros de forma tubular, casi cilíndrica, uniformes y con estrecha distribución de tamaño [33-35]. La presencia de este tipo de histéresis es un indicativo más de que los materiales catalíticos analizados son mesoporosos.
A valores de P/Po comprendidos entre 0.05 y 0.3, la adsorción se realiza sobre las paredes de los poros, dando lugar a la formación de una monocapa. En esta zona, la trayectoria de la isoterma adsorción es igual a la de la isoterma tipo II, por lo que es válido usar la ecuación BET para calcular el área específica de los sólidos [10]. En la Tabla 1 se listan las propiedades texturales del soporte, del catalizador fresco y usado. Como puede observarse, el área específica del soporte es mayor (69.4 m2/g) que la del catalizador impregnado con paladio (57.10 m2/g). Este comportamiento se puede atribuir a la incorporación del metal en el soporte, aunque la disminución en el área específica también puede estar relacionada con deposición de los productos de la oxidación incompleta del acetilacetonato de paladio durante la etapa de calcinación [17,36-40], los cuales se identificaron en los análisis TPR que se mostrarán más adelante.

Igualmente, el área superficial del catalizador usado disminuyó (51.0 m2/g), indicando que el material catalítico perdió área específica durante el proceso de hidrodecloración. Los resultados obtenidos por DRX demuestran que la estructura cristalina de la titania (anatasa) del catalizador usado no cambió durante la reacción de hidrodecloración. Por lo tanto, la disminución del área específica del mismo no está relacionada con transformaciones del soporte. La pérdida de área específica puede ser atribuida a la deposición del NaCl (producto de la reacción) sobre la superficie del catalizador o tal vez, el HCl producido durante la reacción pudo disolver parte del catalizador [41,42].
La Figura 4 presenta los resultados de la distribución de tamaños de poro para el soporte y el catalizador fresco y usado, calculado a partir de los datos de la curva de desorción de las muestras. El procedimiento para realizar los cálculos se tomó de Farrauto y Bartholomew [43]. En la Figura, se observa claramente que el máximo en la distribución de tamaños de poro para todas las muestras analizadas se encuentra en el rango de los mesoporos (5-13 nm), ya que según la clasificación, los poros con tamaños entre 2 y 50 nm, pertenecen a este tipo [34,35,43]. Además, también se puede observar que la mayor contribución de volumen de mesoporos está en 10.40 nm para el soporte y aproximadamente 13.00 nm para el catalizador fresco y usado.

3.4 Espectroscopia de absorción atómica y análisis de quimisorción de hidrógeno
En la Tabla 2 se muestra el contenido de paladio real, la dispersión y el diámetro de partícula para el catalizador fresco y usado. El valor real de paladio obtenido por AAS (0.78 %) es muy similar al contenido metálico teórico (0.80%), es decir, que el método de impregnación húmeda incipiente fue adecuado para depositar la fase activa, ya que se logró depositar casi completamente el paladio de la solución del precursor sobre la superficie del soporte. Además, el valor obtenido para la dispersión; calculada mediante la Ecuación 2, demuestra que el paladio fue bien dispersado sobre el soporte. Según Santamaría [44-46], la incorporación de paladio mediante la impregnación de precursores orgánicos en solventes no acuosos, en términos generales, conduce a una mayor dispersión, por la naturaleza enlazante del anclaje de las partículas de paladio.


Donde:
Pd: porcentaje de dispersión
Vs: volumen adsorbido (cm3 a condiciones estándar)
SFcal : factor estequiométrico
Sw: Peso de la muestra (g)
Fn: fracción o peso de la muestra metálica
Gmwcalc: masa atómica (g/gmol)
Al comparar el contenido de paladio del catalizador fresco con el usado, se observa que la pérdida de metal activo durante las reacciones de hidrodecloración catalítica en fase líquida es mínima. Por otro lado, la dispersión del catalizador usado disminuyó con respecto a la del catalizador fresco. Según algunos estudios [24], en las reacciones de hidrodecloración, las partículas de paladio pueden migrar y aglomerarse, y por lo tanto, el tamaño de las partículas aumentará y como consecuencia, la dispersión disminuirá.
Según Belver at al. [47], el método utilizado para determinar la dispersión (reacciones 2 y 3), puede tener problemas asociados con la formación de hidruros de paladio, lo cual puede resultar en una sobre-estimación de la quimisorción de hidrógeno. La formación de hidruros de paladio a temperaturas menores de los 100°C, puede implicar que la absorción de hidrógeno durante la quimisorción sea mayor que la correspondiente a la estequiométrica, dando lugar a resultados erróneos en la dispersión de paladio y por ende en el tamaño de las partículas. Por esta razón las quimisorciones se realizaron a 100 °C, para evitar la formación de los hidruros de paladio que posteriormente se descompondrán y cuya presencia distorsiona la cantidad de hidrógeno consumida afectando los resultados. El diámetro de las partículas de paladio, ds, en nanómetros, para el catalizador fresco y usado, se estimó a partir de la dispersión del metal, usando la Ecuación 3, en la que se asume que las partículas de paladio son esféricas con una densidad r de 12,02 g/cm3 y un área atómica S de 0,0787 nm2, Mw es el peso atómico del paladio 106,42 g/mol, NA es el número de Avogadro y %D es el porcentaje de dispersión.

Al comparar los valores de los diámetros promedio de las partículas de paladio, determinadas por quimisorción (2.69 nm) y por TEM (2.00 nm), la diferencia es pequeña, lo que permite concluir que las condiciones utilizadas en los análisis de quimisorción de hidrógeno, atenuaron los errores debidos a la formación y posterior descomposición de hidruros de paladio [48,49].
PdO + ¾H2 Û PdH + H2O (Reacción 3)
3.5 Reducción con hidrógeno a temperatura programada (TPR-H2)
Los perfiles de los TPR del soporte y los catalizadores fresco y usado se muestran en la Figura 5. El perfil del TPR del soporte, muestra un pico de reducción entre 600 y 700ºC [11,37]. Al comparar los perfiles de los TPR del catalizador fresco con el usado se puede observar que se obtienen picos de reducción comunes para ambos catalizadores, alrededor de 60, 100, 290 y 400°C. El pico negativo alrededor de 60°C, correspondiente a la liberación de hidrógeno, producto de la descomposición térmica de la fase b-hidruros de paladio (b-PdH), formados a bajas temperaturas durante los TPR [48,49]. La reducción del óxido de paladio, PdO, no soportado es termodinámicamente favorable, pues su energía libre de Gibbs siempre es negativa por encima de -73°C. Aunque el comportamiento de la reducción de los óxidos de metales preciosos puros es diferente a cuando se encuentran soportados, se ha encontrado que la reducción del óxido de paladio soportado también se da a temperaturas relativamente bajas (<100°C). De hecho, en muchas investigaciones, se han identificado grandes picos de consumo de hidrógeno en los perfiles TPR a temperaturas alrededor de 30°C e inclusive temperaturas inferiores que corresponden a la reducción del óxido de paladio [44,48,50]. La reducción del paladio a bajas temperaturas puede suceder según las reacciones 4 y 5, en las cuales el Pd del óxido de paladio al reaccionar con hidrógeno se reduce a hidruro de paladio, el cual según la reacción 6, posteriormente liberará hidrógeno para convertirse en paladio metálico [11,44,50].
1,5 H2 + PddO ® PddH + H2O (Reacción 4)
(1+ 0,5x) H2 + PdbO ® Pdb Hx + H2O (Reacción 5)
Pdb Hx ® Pdb + 0,5x H2 (Reacción 6)

Fagherazzi et al. [51], al realizar estudios estructurales para describir la estequiometría entre el hidrógeno adsorbido y absorbido sobre catalizadores de Pd/SiO2 en función de la dispersión de paladio, encontraron que la concentración de la fase hidruro disminuye con el incremento en la dispersión de paladio. Igualmente, Sá et al. [49], estudiaron los factores que influyen en la formación de hidruros en los catalizadores de Pd/TiO2, encontrando que la concentración de la fase b-hidruro de paladio depende la cantidad y naturaleza de paladio superficial expuesto. Por otro lado, en varias investigaciones se ha reportado que la cantidad de hidrógeno absorbido en la superficie del paladio disminuye con el incremento en la dispersión. Por lo tanto, el aumento en la intensidad del pico de descomposición de la fase b-PdH en el catalizador usado con respecto al fresco, puede ser atribuido al aumento de tamaño de las partículas de paladio (menor dispersión) en el catalizador usado [48-52].
El pico positivo con un máximo a 100°C se ha atribuido a la reducción de especies de PdO superficiales a paladio metálico [40,53-55]. Hosseini at al. [40], identificaron este mismo pico en catalizadores 0,5% Pd/TiO2, debido a la reducción de especies PdO más dispersadas en la estructura porosa. Los picos de reducción con máximos a 290 y 256°C en los catalizadores fresco y usado, respectivamente, se deben probablemente a la reducción de residuos orgánicos [37,52]. Estas sustancias al reducirse consumen hidrógeno y pueden producir algunos hidrocarburos de bajo peso molecular como el metano, el cual fue identificado por MS junto con el etano y el etileno en la corriente proveniente de los TPRs [37,55].
Generalmente, la fijación del acetilacetonato de paladio, Pd(C5H7O2)2, sobre soportes anfóteros como la alúmina o la titania ocurre por un mecanismo de intercambio del ligando. El mecanismo consiste en que el oxígeno de un grupo superficial del soporte, S-OH, forma un enlace covalente con el paladio y se libera la acetilacetona (C5H8O2), que permanece adsorbida en la superficie del soporte.
El proceso de calcinación tiene como objetivo mineralizar completamente los ligandos orgánicos a agua y dióxido de carbono, no obstante, estos ligandos orgánicos o algunos precursores de su descomposición pueden permanecer adsorbidos sobre el soporte, aún después del proceso de calcinación [37,44,38,39,56]. Por ejemplo, Kohler et al. [57], propusieron que los acetilacetonatos metálicos se pueden descomponer parcialmente por la acción catalítica de sitios ácidos y básicos del soporte, formando otros compuestos orgánicos intermediarios como la acetona y el ácido acético, los cuales se adsorben fuertemente en el soporte. Además, Arnoldy y Moulijn [58] mostraron que la reducción de la acetona requiere consumo de H2 dando como resultado la producción de CH4. Similarmente, Aristizábal et al. [37], al analizar los catalizadores Pd/TiO2 sol-gel, identificaron un pico de reducción entre 220 y 280°C aproximadamente, el cual fue atribuido a la reducción de la acetona precursor de la descomposición parcial de los ligandos del acetilacetonato de paladio. Aunque antes de realizar los TPRs, se realizó una oxidación de los catalizadores, vale la pena aclarar que las muestras ya se habían reducido, esto nos permite deducir que los precursores orgánicos (acetilacetona y/o acetona) no fueron eliminados durante el proceso de reducción.
El pico de consumo de hidrógeno a 400°C se ha asignado a la reducción de especies PdO que interactúan fuertemente con el soporte [53,55]. Seshu et al. [53], identificaron el mismo pico de reducción a 400°C en catalizadores de Pd/Al2O3, según ellos este pico se ha atribuido a un complejo superficial 2D (2- dimensional) de PdO. Igualmente, Barrera et al. [59], denominaron este pico de reducción a alta temperatura, [PdO]SC (interacción componente-soporte), el cual se forma por las altas temperaturas de oxidación utilizadas y es más estable a la reducción que el PdO que aparece a bajas temperaturas. Gaspar y Dieguaz [54] han reportado que esta interacción soporte-componente activo se presenta por las altas temperaturas usadas en el proceso de calcinación.
3.6 Espectroscopia de reflectancia difusa de UVVis (UV-Vis DRS)
El análisis por DRS UV-Visible nos puede dar información adicional acerca del estado electrónico del paladio en los catalizadores. La Figura 6 muestra el espectro obtenido para el soporte y el catalizador fresco y usado. En los espectros de las tres muestras se observa una amplia banda entre 200 y 400 nm, la cual se ha atribuido a la naturaleza semiconductora de la titania [30,47]. Al comparar esta banda de absorción (entre 200 y 400 nm) de la titania pura con la del catalizador fresco, se puede observar que se presenta disminución en la intensidad en el catalizador fresco, lo que podría indicar que el paladio cubre una mayor área de la superficie de la titania cuando es impregnado, dejando una fracción considerable del soporte sin interactuar con la radiación. En el catalizador usado, la disminución en la intensidad de dicha banda se atribuye a la adsorción superficial de algunas especies, producto de la reacción como por ejemplo el NaCl.

El leve incremento en la reflectancia entre 380 y 700 nm sugiere la presencia de partículas de paladio metálicas en el catalizador y la inclinación alrededor de 500 nm se ha asignado a las transiciones d-d de las partículas de óxido de paladio [47]. La mayoría de autores afirman que en los espectros UV-Vis de los catalizadores de paladio, las especies Pd0 están asociadas a una absorción sin estructura definida en todo el rango del espectro [60]. No fue posible comprobar la presencia de otras especies Pd2+ en los catalizadores analizados por esta técnica, ya que las bandas asignadas al Pd2+ aparecen entre 300-320 nm y 360-370 nm [54] y son solapadas por la amplia banda de absorción de la titania que apareció entre 200 y 400 nm. Por lo tanto, la técnica no permite obtener mucha información acerca de los estados de oxidación del paladio, especialmente porque el soporte tiene gran capacidad de absorción impidiendo la visualización de algunas bandas importantes.
3.7 Desorción de NH3 a temperatura programada (TPD-NH3)
La acidez superficial de los catalizadores juega un papel importante en el mejoramiento de los procesos catalíticos. La concentración total de los sitios ácidos y la estimación de sus fuerzas relativas, se determinó mediante la desorción de amoníaco con temperatura programada. Con respecto a la fortaleza de los sitios ácidos, algunos autores [61,62] han reportado que las señales de desorción de amoníaco menores de 200°C corresponden a sitios con acidez superficial débil, mientras que las señales de desorción exhibidas entre 200-400°C están asociadas a sitios ácidos de fuerza media, y las señales encontradas a temperaturas mayores de 400°C, corresponden a sitios con acidez fuerte. Es decir, que según las curvas trazadas en la Figura 7, la fuerza ácida de los catalizadores analizados es débil y media, ya que se puede observar que los picos de desorción del NH3 se dan a temperaturas menores de 400°C.

En la Tabla 3 se presenta la acidez total superficial y la máxima temperatura de desorción del amoníaco para el soporte y los catalizadores fresco y usado. Los resultados se obtuvieron a partir del cálculo de las áreas bajo las curvas de desorción de amoníaco y las temperaturas máximas a las cuales se desorbió el amoníaco. Como se puede observar, el catalizador fresco presenta mayor acidez superficial que la titania. Asimismo, la acidez superficial total del catalizador usado es mayor que la del catalizador fresco. La acidez de la titania juega un papel importante en la adsorción de compuestos organoclorados que ocurre en sitios ácidos a través de enlaces entre el cloro y los protones en la superficie, ya que la velocidad de decloración se incrementa con la capacidad del soporte para adsorber el organoclorado [4,11,63].

El aumento de la acidez superficial del catalizador impregnado con paladio se ha explicado por la formación de un tipo de especies Pd-Ti, que aumentan la cantidad de sitios ácidos [64,65]. Según López et al. [62], las mezclas de óxidos metálicos exhiben un gran número de sitios ácidos, que dependen de la cantidad relativa del óxido metálico, la naturaleza y el método de preparación. Tanabe et al. [66], propusieron un modelo basado en la oxidación y en el número de coordinación que involucra los metales para explicar el aumento de la acidez cuando se mezclan dos óxidos: un exceso de carga positiva puede producir acidez Lewis, mientras que un exceso de carga negativa puede ser responsable de la acidez tipo Brönsted. Los análisis de TPR permitieron evidenciar la presencia de especies Pd y PdO en el catalizador, las cuales pudieron ser responsables del aumento de la acidez en el catalizador fresco con respecto al soporte.
En cuanto al aumento de la acidez total superficial del catalizador usado con respecto al fresco, se puede deber al HCl producido durante la reacción de hidrodecloración, ya que el HCl al reaccionar con soportes inorgánicos como la titania, causa un incremento en la acidez superficial, lo que a su vez puede favorecer la formación de depósitos carbonosos en la superficie del catalizador [9,24,64,67]. Según algunos estudios el ácido producido en la reacción conduce a un incremento en la acidez de este material, lo cual a su vez puede llevar además a una redistribución de los sitios activos, incrementando la actividad del catalizador. Hasta donde se pudo investigar, no se encontraron reportes que permitieran diferenciar entre los efectos de la acidez intrínseca y extrínseca del HCl en el soporte de titania. Teniendo en cuenta que en la hidrodecloración siempre va a estar presente el HCl sobre cada soporte, no es posible conocer exactamente el aporte de cada uno, ya que puede haber cierto grado de interacción entre ellos, favoreciendo la reacción de hidrodecloración. Lo que si se ha reportado es que el HCl actúa sobre el paladio, desactivándolo [11].
La presencia de sitios ácidos en los catalizadores analizados es una característica más del catalizador que probablemente favoreció la actividad catalítica en la reacción de hidrodecloración. Wook et al. [68], al realizar la hidrodecloración del CCl4 con catalizadores de Pt y Pd soportados, obtuvieron las conversiones y selectividades más altas con los catalizadores de SiO2 y Al2O3, los cuales presentaban mayor acidez superficial.
3.8 Espectroscopia infrarroja (IR) de NH3 y CO adsorbidos
En general la acidez superficial de las titanias es de tipo Lewis y ha sido caracterizada esencialmente por adsorción de amoníaco, sin embargo, datos recientes [69] demuestran que el amoníaco no es una molécula prueba lo suficientemente sensitiva para distinguir entre los diferentes tipos de sitos ácidos Lewis en la titania anatasa, por lo tanto, es recomendable hacer un estudio complementario con CO adsorbido, ya que el CO ha sido la molécula prueba más sensitiva para la determinación de la fuerza de sitios ácidos no protónicos y de sitios ácidos Lewis de diferente carácter energético. Además, el análisis por espectroscopia infrarroja de CO adsorbido también permite identificar los estados electrónicos del metal soportado [30,69-71].
En la Figura 8 se muestran los espectros IR de amoníaco adsorbido en el soporte y en los catalizadores fresco y usado. No se observaron bandas típicas en la región de altos números de onda, es decir, la superficie de los catalizadores usados en el presente trabajo está pobremente hidroxilada. Se observaron picos característicos a 1177, 1300, 1465, 1600 y 1673 cm-1, la primera y tercera bandas corresponden a la adsorción de NH3 sobre sitios ácidos Lewis. La señal a 1300 cm-1 se ha asignado a formas de amonio disociadas. Por último, el pico de adsorción a 1600 cm-1, caracteriza las vibraciones asimétricas de amoníaco enlazado coordinadamente (das(NH3)), no obstante, esta banda también puede corresponder a los grupos OH del agua adsorbida. La señal débil a 1673 cm-1 se ha atribuido a los modos de deformación simétrica de iones NH4+ sobre sitios ácidos Brönsted, la identificación en estos sitios se puede deber a la presencia de ciertas impurezas en el catalizador, las cuales pueden producir este tipo de acidez [69,72,73].

La banda de absorción que aparece como un doblete en el soporte (Figura 8a, 1177 y 1200 cm-1) corresponde a vibraciones simétricas (dsy(NH3)) del amoníaco adsorbido coordinadamente. Según Watson y Ozkan [72], esta banda está localizada sobre sitios ácidos Lewis tipo b, los cuales son sitios Ti4+ pentacoordinados. La señal hallada a 1465 cm-1 es típica de iones amonio (das(NH4+)), los cuales fueron identificados en la superficie de la titania anatasa en un estudio realizado por Hadjiivanov et al. [69]. No se observan señales a 1421, 1437 y 1673 cm-1 ubicadas en la región de bajos números de onda, características de la adsorción de NH3 sobre sitios ácidos Brönsted [69,70]. Los resultados obtenidos son coherentes con lo reportado anteriormente, en donde se afirma que la titania anatasa generalmente posee baja acidez tipo Brönsted.
En la Figura 9 se muestra el espectro de adsorción de CO sobre el soporte a temperatura ambiente. La señal débil a 1625 cm-1 está relacionada con el modo de alargamiento de los grupos hidroxilos del agua (H-O-H) [69]. La banda a 2127 cm-1 corresponde al CO físicamente adsorbido, y la última banda a 2186 cm-1 se ha asignado a grupos carbonilos adsorbidos en sitios ácidos Lewis b-Ti4+ y corresponde a la formación de complejos Ti4+-CO. Esta misma señal también se ha identificado en los espectros de CO adsorbido sobre TiO2 reducida, por lo tanto, la banda también se ha asignado al monóxido de carbono coordinado a sitios b-Ti3+ [30,69,70].

Casi todos los autores que han estudiado la adsorción del CO sobre la titania anatasa a temperatura ambiente, han reportado la existencia de solamente dos tipos de sitios Lewis con entalpía de adsorción diferente (sitios a y sitios b). La adsorción del CO sobre sitios altamente energéticos (sitios a) está caracterizada por una banda a 2208 cm-1 o 2206 cm-1 y la adsorción del CO en sitios moderadamente energéticos (sitios b) produce una banda de adsorción a 2192 cm-1 o 2186 cm-1. Hay un consenso general en que los sitios ácidos Lewis en la anatasa detectados por adsorción de CO tienen coordinación de 4 y 5 con cationes insaturados Ti4+, los cuales están situados en las diferentes caras (picos, esquinas, lados) de los cristales del óxido de titanio [30,47,69,70].
Aunque en la literatura, se ha evidenciado la presencia de una gran variedad de sitios ácidos Lewis (a, b, g) con diferente fuerza ácida en la superficie de las titanias [19,39,71-73], los resultados de espectroscopía IR, sugieren que los catalizadores analizados en el presente trabajo poseen una superficie débilmente hidroxilada con acidez tipo Lewis y contribución básicamente de sitios b, los cuales son moderadamente energéticos. Estas observaciones son consistentes con los resultados obtenidos en los TPD de amoníaco.
El buen desempeño de la titania en las reacciones de hidrodecloración se ha atribuido a la acidez superficial (muchas reacciones son catalizadas por sitios ácidos en la superficie del catalizador). La eliminación de grupos OH superficiales en forma de agua hace que los átomos superficiales de la titania se comporten como ácidos Lewis. Los sitios ácidos Lewis se caracterizan por la ausencia de grupos hidroxilos en los átomos de titanio, los cuales al estar positivamente cargados poseen una mayor habilidad para capturar electrones y pueden adsorber con mayor fuerza los compuestos organoclorados. Consecuentemente, los centros BrÖnsted son muy escasos, si bien su acidez puede potenciarse mediante la incorporación de halógenos (cloruros) o sulfatos, en la estructura del mismo. En el tratamiento de organoclorados los átomos de cloro liberados pueden producir precisamente este efecto, mediante la adsorción del compuesto organoclorado sobre los centros ácidos, por medio de un enlace de hidrógeno entre el cloro y el protón superficial [24,29,64,65,74-76].
La espectroscopía IR de CO adsorbido también es un método usualmente utilizado para la caracterización de los estados electrónicos de los metales soportados. De acuerdo con el modelo de enlace s-p reportado por Davydov, las moléculas de monóxido de carbono adsorbidas sobre los átomos o iones del metal interactúan con sus electrones d de valencia y la frecuencia de las vibraciones de alargamiento del enlace C-O (nCO) de los complejos superficiales M-CO depende directamente de la carga efectiva del adsorbente. La interpretación de las bandas de adsorción del CO sobre los catalizadores de paladio soportado está bien definida en la literatura. Las señales en el intervalo 1850- 1980 cm-1 y 2050-2100 cm-1 son atribuidas al puente tendido Pd-CO-Pd y a las especies Pd0-CO, las bandas a 2120-2130 y 2150-2160 cm-1 corresponden a los complejos Pd+-CO y Pd2+-CO, respectivamente [19,39,71,77-78].
En la Figura 9 se observa que los espectros IR de CO adsorbido para el catalizador fresco y usado son muy similares. Estos espectros mostraron bandas características a 1628, 1930, 1980, 2100, 2131 y 2183 cm-1. La banda a 1628 cm-1 se asigna al modo asimétrico de especies de bicarbonato (nas(CO)) adsorbidas en la superficie de los grupos OH del agua, la cual fue detectada anteriormente. La banda a 1930 cm-1 y el hombro a la derecha de este pico ubicado a 1980 cm-1, son atribuidos a una molécula de CO enlazada a dos centros de paladio metálico [(Pd0)2-CO (iso)]. La señal a 2100 cm-1 corresponde a especies carbonilos (CO) coordinadas con partículas metálicas de paladio (Pd0- CO lineal), aunque esta banda se solapa con la banda que aparece en la misma región en el soporte (2130 cm- 1), el aumento significativo en la intensidad de este pico en el soporte impregnado con paladio comprueba la presencia de partículas de Pd metálico en el catalizador. El hombro que aparece a la derecha de este pico a 2131 cm-1, indica la presencia de especies de Pd2+-CO, comúnmente asociadas con especies PdO formadas por la oxidación de los ligandos del acetilacetonato de paladio. La última banda que aparece a 2183 cm-1 identificada en los anteriores espectros, corresponde a grupos carbonilos formados en sitios ácidos Lewis tipo b [19,30,39,47].
3.9 Análisis termogravimétrico (TGA-DTG)
En las Figuras 10, 11 y 12 se muestran las curvas de los análisis termogravimétricos (TGA) con sus respectivas derivadas (DTG) realizados al soporte, el catalizador fresco y usado, respectivamente. En el termograma del soporte y de los catalizadores fresco y usado, se observan picos a temperaturas inferiores de 160°C, esto corresponde a la pérdida de agua físicamente adsorbida u ocluida en el catalizador, y a la remoción de solvente físicamente adsorbido durante la etapa de preparación del soporte y del catalizador [10,11,24,67]. La ausencia de otros picos diferentes en el soporte sugiere que éste no presenta depósitos carbonosos u otros compuestos orgánicos, lo cual concuerda con los resultados de TPR.



En los termogramas de los catalizadores fresco y usado, se observan dos picos de combustión que no están presentes en el soporte. Los intervalos de temperatura en los que aparecen estos dos picos para el catalizador fresco y usado son 160-250°C y 270- 402°C, respectivamente, es decir, son muy similares para ambos catalizadores. Marécot et al. [79], establecieron temperaturas de oxidación para depósitos carbonosos sobre catalizadores de paladio soportados en cualquier soporte entre 200 y 600°C. De acuerdo con esto, la pérdida de peso en los intervalos de temperatura anteriores (160-402°C) se atribuye a la combustión de residuos orgánicos, los cuales pueden estar asociados a sitios ácidos del soporte o a la fase metálica del catalizador. Los valores de pérdida total de peso en porcentaje (D%) y los intervalos de las temperaturas de los principales picos de combustión se muestran en la Tabla 4. Para este tipo de catalizadores se asume que los reactivos son desorbidos a temperaturas inferiores de 200°C y que la pérdida de peso es insignificante a temperaturas mayores de 800°C.

Aunque en el catalizador usado, no se observan picos de combustión diferentes a los presentes en el catalizador fresco, la pérdida de peso total respecto a la cantidad de muestra inicial para el catalizador usado (1.73%) fue un poco mayor que para el fresco (1.44%), lo cual sugiere que durante la reacción algunas sustancias orgánicas se pudieron adsorber en la superficie del catalizador.
3.10 Microscopia de transmisión electrónica (TEM)
En las Figuras 13a-13c se muestran los resultados de las micrografías obtenidas por TEM para el soporte, el catalizador fresco y usado. Como la microestructura del soporte no permitió obtener un contraste de tamaño atómico suficiente para diferenciar las partículas de paladio de los cristales de titania, fue necesario complementar el estudio con la indización cristalográfica de un patrón de difracción de electrones para identificar las partículas de paladio. Para determinar el patrón de difracción característico del paladio se utilizó como estándar de calibración una película de oro.

Las micrografías TEM del soporte revelan la presencia de agregados de poliedros irregulares compuestos de partículas elementales con un tamaño en un intervalo entre 28.4 y 45.4 nm. Tal geometría de la fase anatasa es consistente con los resultados obtenidos en otros estudios de titania solgel [28] además, el tamaño promedio de los cristales es muy cercano al determinado con la fórmula de Scherrer (Tabla 5).

La morfología típica de los cristales de paladio es pseudoesférica. Al comparar las micrografías en campo oscuro del catalizador fresco con el usado, se puede observar que no hay modificaciones apreciables en su morfología, sin embargo, se observa un incremento en sus tamaños (menor dispersión) en el catalizador usado debido probablemente a la aglomeración de los cristales durante el curso de la reacción, este resultado puede ser explicado por efecto del HCl, el cual puede favorecer la coalescencia de los cristales [24,80]. Esto confirma los resultados obtenidos en los análisis de quimisorción de hidrógeno.
4. CONCLUSIONES
Los resultados de caracterización de los catalizadores frescos, revelaron la obtención de titania anatasa (89.4%), conformada principalmente de agregados de poliedros irregulares compuestos de partículas elementales con un tamaño promedio de 38.9 nm. El material mesoporoso con un área especifica relativamente alta (69.4 m2/g) y débilmente hidroxilado, presenta acidez tipo Lewis con contribución básicamente de sitios b, los cuales son moderadamente energéticos, además presente algunos residuos orgánicos a nivel superficial. El método de impregnación húmeda incipiente y los métodos de tratamiento térmico y reductivo aplicados a los catalizadores, permitió obtener materiales con un contenido de paladio de 0.778% w.w, moderadamente dispersos (42.804%), con especies de Pd0 y Pd2+ en la superficie y cuyas partículas en forma seudoesférica presentan un diámetro promedio de 2.69 nm aproximadamente. No obstante, la incorporación del metal en el soporte produjo una disminución del 26.5% del área específica y un ligero aumento de la acidez Lewis.
Al comparar los resultados de caracterización de los catalizadores frescos con los de los catalizadores usados en las reacciones de hidrodecloración de tetracloroetileno en fase líquida, se revelan ciertos cambios como: disminución en el área especifica, aumento de la acidez superficial, presencia de algunos compuestos orgánicos, disminución de la dispersión y con ello el aumento en el tamaño de las partículas de paladio. No obstante, algunas características como la estructura cristalina del soporte, el contenido metálico, la forma de las partículas de paladio y sus estados de oxidación permanecieron casi constantes. El factor que más contribuyó al cambio en las características de los catalizadores se atribuye principalmente al valor del pH del medio producido por el cloruro de hidrógeno formado durante la reacción y al exceso de hidróxido de sodio adicionado a la mezcla de la reacción para neutralizar dicho ácido.
Los resultados obtenidos en las caracterizaciones realizadas a los catalizadores usados, permite concluir que en general los materiales sintetizados son estables en las reacciones de hidrodecloración catalítica de tetracloroetileno en fase líquida bajo las condiciones aplicadas, ya que conservan muchas de sus características iniciales. Con esto se confirma que estos catalizadores son realmente promisorios para el tratamiento y desinfección de aguas contaminadas con solventes organoclorados ligeros.
5. REFERENCIAS BIBLIOGRÁFICAS
1. Ardila A. A. Hidrodecloración de mezclas mono y multicomponentes de organoclorados ligeros en fase líquida con catalizadores de paladio soportados sobre titania preparada por el método sol-gel. Tesis MSc. Medellín (Colombia): Universidad de Antioquia, 2008. [ Links ]
2. Mackenzie K., Frenzel H., Kopinke F. Applied Catalysis B: Environmental. 2006; 63: 161-167. [ Links ]
3. Padilla B.V., Díez S. F., Ordóñez G.S. Tratamiento de efluentes acuosos contaminados con compuestos organoclorados. En: Ingeniería del agua. 2005; 12, p. 361-375. [ Links ]
4. S. Ordóñez. Hidrodecloración catalítica de compuestos alifáticos clorados, Tesis Ph.D. Oviedo (España): Universidad de Oviedo, 2000. [ Links ]
5. Alonso F., Beletskaya I.P., Yus M. Chemical Review. 2002; 102, 4009-4091. [ Links ]
6. Kim D., Allen D.T. Engineering Chemical Research., 1997; 36, 3019- 3026. [ Links ]
7. Centi B. Journal of Molecular Catalysis A: Chemical., 2001; 173, 287-312. [ Links ]
8. Martino M., Rosal R., Sastre H, Díez F.V. Applied Catalysis B: Environmental., 1999; 20, 301-307. [ Links ]
9. Ordóñez S., Sastre H., Díez F.V. Applied Catalysis B: Environmental., 2003; 40, 119-130. [ Links ]
10. B. H. Aristizábal, Hidrodecloración catalítica en fase gaseosa de compuestos organoclorados sobre catalizadores sol-gel, Tesis MSc. Medellín (Colombia): Universidad de Antioquia, 2003. [ Links ]
11. González A. Hidrodecloración catalítica de diclorometano en presencia de cloroformo y/o tetracloroetileno: Estudio cinético. Tesis MSc. Medellín (Colombia): Universidad de Antioquia, 2006. [ Links ]
12. Aristizabal B., González C. A, Barrio I., Montes M., Montes de Correa C. Journal of Molecular Catalysis A: Chemical., 2004; 222, 189-198. [ Links ]
13. Martínez L., Montes C., Odriozola J., Centeno M. Journal of Molecular Catalysis A: Chemical., 2006; 253, 252-260. [ Links ]
14. González A., Bustamante F., Montes de Correa Consuelo. Hidrodecloración catalítica de diclormetano, cloroformo y tetraclorotetileno. En: Revista Facultad de Ingeniería. 2006, 38, p. 73-87. [ Links ]
15. Malinowski A., Lomot D., Karpinski Z. Applied Catalysis B: Environmental., 1998; 19, L79-L86. [ Links ]
16. Sai Prasad P.S. Catalysis Letters., 2000; 66, 201–204. [ Links ]
17. Krishnankutty N., Li J., Vannice M. Applied catalysis A: General., 1998; 173, 137-144. [ Links ]
18. Toebes M., Dillen J., Jong K. Journal of Molecular Catalysis A: Chemical., 2001; 171, 75-98. [ Links ]
19. Yuan G., Keane M.A. Journal of Catalysis., 2004; 225, 510-522. [ Links ]
20. Angel D., Benítez J. Journal of Molecular Catalysis A: Chemical., 2001; 165, 9-13. [ Links ]
21. Kopinke F. D., Mackenzie K., Koehler R., Georgi Anett. Applied Catalysis A: General., 2004; 271, 119-128. [ Links ]
22. Ardila A. Alba Nelly, Martínez Miguel A., Zurita Gustavo F., Montes de Correa Consuelo. Hidrodecloración catalítica de percloroetileno en fase líquida. En: Revista Facultad Ingeniería. 2007, 42. p. 132-147. [ Links ]
23. Li Y., Fan Y., Yang H., Xu B., Feng L., Yang M., Chen Y. Chemical Physics Letters., 2003; 372, 160-165. [ Links ]
24. López E., Ordóñez S., Díez F. Applied Catalysis. B: Environmental., 2006; 62, 57-65. [ Links ]
25. Wu W., Xu J., Ohnishi R. Applied Catalysis B: Environmental., 2005; 60, 129-137. [ Links ]
26. S. Ordoñez, F. V. Diez, H. Sastre. Destrucción de compuestos organoclorados mediante hidrogenación catalítica. En: Memorias de las Primeras Jornadas Iberoamericanas de Catálisis Ambiental, CYTED. 2000, p. 277. [ Links ]
27. Aramendía M.A., Borau V., García I.M., Jiménez C., Marinas J.M., Urbano F.J. Applied Catalysis B: Environmental, 1999; 20, 101±110. [ Links ]
28. Khalil Kamal M.S., Baird Thomas, Zaki Mohamed I., E1-samahy Ahmed A., Awad Aida M. Physicochemical and Engineering Aspects, 1998; 132, 31-44. [ Links ]
29. M. Rosa-Brussin. Procesos para reducir las emisiones de compuestos orgánicos volátiles (COVs). En: Programa Iberoamericano de Ciencia y Tecnología para el Desarrollo CYTE. 1998, p. 37-42. [ Links ]
30. Su C., Hong B. Y., Tseng C.M. Catalysis Today, 2004; 96, 119-126. [ Links ]
31. De Farias Robson F. Journal of Colloid and Interface Science, 2001; 239, 584-586. [ Links ]
32. Seilfert J., Emig G. Chemical Engineering, 1991; 31, 29-40. [ Links ]
33. Leofanti G., Padovan M., Tozzola G., Venturelli B. Catalysis Today, 1998; 41, 207-219. [ Links ]
34. Storck S., et. al. Applied Catalysis A., 1998; 174, 137-146. [ Links ]
35. Sing K. Advances in Colloid and Interface Science, 1998; 76-77, 3-11. [ Links ]
36. Panpranot Joongjai, Kunyaluck Kontapakdee and Piyasan Praserthdam. Journal Physics Chemical B, 2006; 110, 8019-8024. [ Links ]
37. Di Paola A., Garcia-López E., Marci G., Martín C., Palmisano L., Rives V., Venezia A. Applied Catalysis B: Environmental, 2004; 48, 223-233. [ Links ]
38. Daniell W., Landes H., Fouad N.E., Knözinger H. Journal of Molecular Catalysis A: Chemical, 2002; 178, 211–218. [ Links ]
39. Santacesaria E. Catalysis Today, 1999; 52, 113-123. [ Links ]
40. Hosseini M., Siffert S., Tidahy H., Cousin R., Lamonier J., Aboukais A., Vantomme A., Roussel M., Su B. Catalysis Today, 2007; 122, 391-396. [ Links ]
41. Aramendía M.A., Boráu V., García I.M, Jimènez C., Lafont F., Marinas A., Marinas J. M., Urbano F.J. Journal of Molecular Catalysis A: Chemical., 2002; 184, 237-245. [ Links ]
42. Ukisu Y., Miyadera T. Applied Catalysis A: General., 2004; 271, 165-170. [ Links ]
43. Farrauto R. J., Bartolome C.H. Blackie Academic & Professional., 1997; 64-66, 117-198. [ Links ]
44. Gonzalez R. D., López T., Gómez R. Catalysis Today., 1997; 35, 293-317. [ Links ]
45. Aramendía M.A., Burch R., García I.M., Marinas A., Marinas J.M, Southward B., Urbano F.J. Applied Catalysis B: Environmental., 2001; 31, 163-171. [ Links ]
46. Kalevaru V., Benhmid A., Radnik J., Lûcke B., Martín A. Journal of Catalysis., 2006; 243, 25-35. [ Links ]
47. Belver C., López-Muñoz M. J., Coronado J. M., Soria J. Appl. Catal. B. Environ. 2003; 46, 497-509. [ Links ]
48. Nag Nabin K A. Journal Physics Chemical., 2001; 105, 5945-5949. [ Links ]
49. Sà J., Arteaga G., Daley R., Bernardi J., Anderson J. Journal Physics Chemical B., 2006; 110, 17090-17095. [ Links ]
50. Wang C., Lin H., Ho C. Journal of Molecular Catalysis A. Chemical., 2002; 180, 285-291. [ Links ]
51. Fagherazzi G., Benedetti A., Polizzi S., Mario A., Pinna F., Signoretto M. y Pernicone N. Catalysis Letter., 1995; 32, 293-303. [ Links ]
52. Lomot D., Juszczyk W., Karpínki Z., Bozon-Verduraz F. Journal Chemical Society Faraday Transition., 1997; 93 (10), 2015-2021. [ Links ]
53. Babu N., Lingaiah N., Gopinath R., Reddy P., Prasad P.S. Journal of Physics Chemical C., 2007; 111, 6447-6453. [ Links ]
54. Gaspar A., Dieguez L. Applied Catalysis A: General., 2000; 201, 241-251. [ Links ]
55. Lieske H., Vólter J. Journal of Physical Chemistry., 1985; 89, 1-2. [ Links ]
56. Campostrini R., Ischia M, Palmesano L. Journal of Thermal Analysis and Calorimetry., 2003; 71, 997-1009. [ Links ]
57. Kohler S., Reiche M., Frobel C.M. Elsevier Amsterdam., 1995; 91, 1009. [ Links ]
58. Arnoldy P., Moulijn J.A. Journal of Catalysis., 1985; 93, 38-54. [ Links ]
59. Barrera A., Viniegra M., Fuentes S., Díaz G. Applied Catalysis B: Environmental., 2005; 56, 279-288. [ Links ]
60. Pestryakov A. N., Lunin V. V., Fuentes S., Bogdanchikova N., Barrera A. Chemical Physics Setter. 2003; 367, 102-108. [ Links ]
61. J. L. Agudelo, L. Martínez, Oxidación de trazas de clorobenceno con catalizadores de paladio y níquel soportados en circonia sulfatada sintetizados por el método sol-gel, Tesis MSc. 2003, Medellín (Colombia): Universidad de Antioquia. [ Links ]
62. Li Y., Xu B., Fan Y., Feng N., Qiu A., Jie He J., Yang H., Chen Y. Journal of Molecular Catalysis A: Chemical., 2004; 216, 107. [ Links ]
63. Weber R., Hagenmaier H. Chemosphere. 1999; 38, 529-549. [ Links ]
64. Lomot D., Juszczyk W., Karpinski Z. Applied Catalysis A: General., 1997; 155, 99-113. [ Links ]
65. Skotak M., Karpinski Z., Juszczyk V., Pielaszek J., Kepinski L., Kazachkin D., Kovalchuk V., d Itri J. Journal of Catalysis., 2004; 227, 11-25. [ Links ]
66. K. Tanabe., M. Misono, Y Ono, H Hattori. Tokio Kondansha, Academic Press, 1990. [ Links ]
67. Nishijima W., Ochi Y., Tsai T., Nakano Y., Okada M. AppIied Catalysis B., 2004; 51, 135-140. [ Links ]
68. Kopinke F.D., Mackenzie K., Köhler R. Applied Catalysis B: Environmental, 2003; 44, 2003, 15-24. [ Links ]
69. Hadjiivanov Konstantin. Applied Surface Science., 1998; 135, 331–338. [ Links ]
70. Faccin F., Guedes F., Benvenutti E., Moro C. Ecléctica Química., 2002; 27, 1-10. [ Links ]
71. Cao Y. C., Yiang X. Z., Song W. H., Bai Z. Q., Fang X.Q. Catalysis Letters. 2001; 76, 53-58. [ Links ]
72. Watson J., Ozkan U. Journal of Molecular Catalysis A: Chemical., 2003; 192, 79-91. [ Links ]
73. Hinz A., Larsson P., Skárman B., Andersson A. Applied Catalysis B: Environmental., 2001; 34, 161-178. [ Links ]
74. F. Delannay, B. Delmon. Methods of catalysts characterization an overview. Characterization of heterogeneuos catalyst, capítulo I. Editorial Dekkev. New York, 1998. [ Links ]
75. Shin E. J., Keane M.A. Reaction Kinectics and Catalysis Letters., 2000; 69, 3-8. [ Links ]
76. Benvenutti Edilson V., Franken Leonardo, Moro Celso C., Davanzo Celso U. Langmuir. 1999; 15, 8140-8146. [ Links ]
77. Bae J. W., Jang E. J., Hyun Jo. D., Lee J.S., Lee K.H. Journal of Molecular Catalysis A: Chemical., 2003; 206, 225-238. [ Links ]
78. ATSDR. Biennial Report to Congress 1991 and 1992. Agency for Toxic Substances and Disease Registry. US Department of Health and Human Services, Atlanta, Georgia, 1996. [ Links ]
79. Marécot P., Akhachane A., Micheaud C., Barbier J. Applied Catalysis A : Chemical., 1998 ; 169, 189. [ Links ]
80. Datye A., Xu Q., Kharas K., McCarty J. Catalysis today., 2006; 111, 59-67. [ Links ]
