










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Facultad de Agronomía
versión impresa ISSN 0378-7818
Rev. Fac. Agron. v.25 n.3 Caracas sep. 2008
Optimización y validación de un método de extracción y limpieza en fase sólida para la determinación de insecticidas organofosforados en cebollín (Allium fistulosum L.) y cilantro (Coriandrum sativum L.)
Extraction and cleanup method optimization and validation in solid phase for determining organophosphorus insecticides on green onion (Allium fistulosum L.) and coriander (Coriandrum sativum L.)
G. Ettiene1*, S. Ortega2, D. Medina1, J. Sepúlveda3 y L. Sandoval4
1*Autor de correspondencia e-mail: gettiene@yahoo.com
1Departamento de Química. Facultad de Agronomía, Universidad del Zulia. Venezuela
2División de Extensión. Facultad de Agronomía, Universidad del Zulia. Venezuela
4Instituto de Investigaciones Agronómicas. Facultad de Agronomía, Universidad del Zulia. Venezuela
3MSc. en Ciencias Ambientales. Universidad del Zulia. Venezuela
Resumen
Se optimizó y validó un método analítico para la extracción y limpieza de residuos de insecticidas organofosforados (OPs: diazinón, malatión, paratión, disulfotón, fenitrotión, fenclorfos, clorfenvinfos, y metidatión) en cebollín (Allium fistulosum L.) y cilantro (Coriandrum sativum L.). Se calcularon los porcentajes de recuperación de los OPs para evaluar la eficiencia del método, adicionando a 4g de muestra de cada matriz, libre de residuos de insecticidas, una concentración conocida de los OPs (0,2-10 µg.g-1). La cuantificación se realizó empleando cromatografía de gases capilar (CGC) con detección selectiva nitrógeno fósforo. La fase sólida carbón activado/florisil (200:700mg), permitió la obtención de altos porcentajes de recuperación (96-105%) y extractos limpios. El método de extracción propuesto mostró una alta precisión en términos de repetibilidad (RSD <3%) y reproducibilidad (RSD<7%) y bajos límites de detección (0,0061-0,020 µg.g-1), pudiendo ser empleado para el monitoreo rutinario de niveles residuales de OPs en los materiales vegetales evaluados.
Palabras clave: Insecticidas organofosforados, métodos de extracción, extracción en fase sólida, residuos de insecticidas.
Abstract
An analytical method for extraction and cleanup of organophosphorus pesticides residues (OPs: diazinon, Malathion, parathion, disulfoton, fenitrothion, fenchlorphos, chlorfenvinphos, and methidathion) in green onion (Allium fistulosum L.) and coriander (Coriandrum sativum L.) was validated and optimized. The percentage of recovery of the OPs were calculated to evaluate the efficiency of the method, adding to 4g of sample of each matrix, free of insecticide residues, a known concentration of the OPs (0.2-10 µg.g-1). The quantification was carried out employing capillary gas chromatography (GCC) with selective detection nitrogen phosphorus. The solid phase activated charcoal/florisil (200:700mg), allowed the obtaining of highly percentages of recovery (96-105%) and clean extracts. The extraction method proposed showed a high precision in terms of repeatability (RSD <3%) and reproducibility (RSD<7%), and low detection limits (0.006-0.020 µg.g-1), could be used for the routine monitoring of residual levels of OPs in the vegetable materials evaluated here in.
Key words: Organophosphorus insecticides, extraction methods, solid phase extraction, insecticides residues.
Recibido el 28-5-2007 Aceptado el 5-6-2008
Introducción
Los insecticidas han proporcionado beneficios a la agricultura porque ha permitido controlar y eliminar las plagas y enfermedades que atacan y afectan a plantaciones agrícolas tales como frutales y hortalizas; sin embargo, se ha demostrado que su empleo intensivo e incontrolado constituye un alto riesgo para la salud humana debido a su alta toxicidad directa y residual (Buscema et al., 2004; Calonge, 2002; Danis et al., 2002; Ettiene et al., 1997).
El uso excesivo e indiscriminado de estas sustancias, puede ocasionar que niveles no inocuos permanezcan en los cultivos hasta el momento de la cosecha, e incluso, hasta la postcosecha, lo cual conlleva un alto riesgo de contaminación de los consumidores. Esto implica que el riesgo de intoxicación o envenenamiento sea muy alto, sobre todo cuando se trata de productos que se consumen frescos y sin tratamiento tecnológico, como es el caso de muchas frutas y hortalizas (Calonge, 2002; Danis et al., 2002; Prieto et al., 2002; Sánchez et al., 2005; Schenck y Lehotay, 2000).
La determinación de niveles residuales de OPs requiere disponer de métodos analíticos sensibles que permitan cuantificar niveles trazas de concentración (mg.kg-1), y diseñar propuestas de extracción analíticas como etapas preliminares de tratamiento de muestra. En la actualidad, no existe una sola secuencia de extracción que se pueda aplicar a todo tipo de OPs. La elección de la etapa preliminar depende de la naturaleza química de los componentes y de los sustratos en los que se encuentran (Picó y Font, 2003; Stan, 2000).
La extracción líquido-líquido es una técnica universal de gran aplicación para la extracción de insecticidas de matrices complejas que proporciona excelentes resultados. Sin embargo, el proceso es largo y tedioso y presenta ciertos inconvenientes tales como el empleo de grandes cantidades de disolvente cuyo costo es muy elevado (Itoh et al., 1996; Mol et al., 2003; Tang, 2005).
La extracción en fase sólida (EFS) es una técnica muy utilizada para la extracción de OPs (kadennczki et al., 1992), que además de permitir el empleo de menores cantidades de solventes, evita otros inconvenientes asociados a la extracción líquido-líquido, tales como la separación incompleta de las fases y las interferencias de la matriz. Por otra parte, se obtienen ventajas al permitir la reducción de la cantidad de muestra y del tiempo de extracción.
Las técnicas analíticas comúnmente utilizadas para la determinación de los niveles residuales de OPs son cromatografía líquida (CL) y cromatografía de gases (CG). La mayor parte de los métodos de análisis emplean una variedad de técnicas de detección, entre ellas se han publicado aplicaciones con sistemas de detección nitrógeno fósforo (DNP) (Lucio et al., 2004; Oliva et al., 2000; Prieto et al., 1999; Prieto et al., 2002), detección fotométrica a la llama (DFLL) (Calonge, 2002; Tang et al., 2005) y espectrometría de masas (EM) (Itoh et al., 1996). De éstas, la cromatografía de gases acoplada al detector de masas (CG-EM) y la CG acoplada al detector nitrógeno-fósforo (CGDNP) constituyen las técnicas analíticas más empleadas en la determinación de OPs en diferentes tipos de frutas y vegetales, proporcionando resultados altamente confiables (Calonge, 2002; Prieto et al., 1999; Prieto et al., 2002; Tang et al., 2005).
El objetivo de este trabajo fue optimizar y validar un método de extracción y limpieza de residuos de los OPs (diazinon, malatión, paratión, disulfotón, fenitrotión, fenclorfos, clorfenvinfos y metidatión) en cebollín (Allium fistulosum L.) y cilantro (Coriandrum sativum L.).
Materiales y métodos
La investigación se desarrolló en la Sección de Cromatografía del Instituto de Investigaciones Agronómicas de la Facultad de Agronomía, Universidad del Zulia, Maracaibo.
Estándares de OPs de alta pureza de diazinón (98,9%), disulfotón (97,09%), fenclorfos (99,1%), fenitrotión (97,0%), malatión (98,5%), paratión (99,2%), clorfervinfos (95,0%) metidatión (97,7%), (Dr. Ehrenstorfer GMBHâ, Alemania), se utilizaron para preparar, soluciones madre (1000 µg.mL-1) de cada insecticida en acetato de etilo grado HPLC (Bakerâ, USA), a partir de las cuales se prepararon por dilución soluciones de calibración en acetato de etilo grado HPLC (Bakerâ, USA) y soluciones para adición estándar en metanol. (Bakerâ, USA).
Se preparó una solución madre (1000 µg.mL-1) del estándar interno trifenilfosfato (TPP) (99%) (Riedel de Haënâ, Alemania), en acetato de etilo. Todas las soluciones se protegieron de la luz con papel de aluminio y se almacenaron bajo refrigeración (4ºC).
Carbón Grafitado (120/400 mesh, CarboGraphâ, Alltech), Carbón Activado (6-60, Fisher Scientific, USA) y Florisil (Riedel de Haënâ, Alemania) 0,15020,25 mm (60-10 mesh Astm) se utilizaron como fases sólidas en los ensayos para la limpieza de los extractos de los materiales vegetales utilizados (cebollín y cilantro).
Los análisis se realizaron empleando un Cromatógrafo de Gas, modelo Auto System, Perkin-Elmer, equipado con un detector termoiónico (Nitrógeno-Fósforo), un muestreador Automático Perkin-Elmer, una columna capilar (30 m x 0,53 mm x 1,2 µm de espesor de película, de 5% fenil 95% metil silicona (AT-5, ALLTECH, USA). El registro de los cromatogramas y la integración del área de los picos se realizaron en un computador, equipado con un soft ware Tubochrom Navigator versión 4,1 (Perkin-Elmer).
Optimización de la separación cromatográfica
La optimización de la separación cromatográfica de los OPs se realizó empleando las condiciones reportadas por Ettiene et al., (1997). Las curvas de calibración por el método del estándar interno se obtuvieron por inyección de soluciones patrón (0,25-4,0 µg.mL-1) conteniendo todos los OPs y el estándar interno.
Los límites de detección (Miller-Miller, 1993) se determinaron inyectando por triplicado soluciones patrón de los OPs (0,025-0,1 µg.mL-1).
Optimización del método de extracción y limpieza
La etapa de optimización de la extracción de los OPs en cebollín (Allium fistulosum L.) y cilantro (Coriandrum sativum L.), constituyó la fase más importante de este trabajo por cuanto se debió cumplir simultáneamente con dos objetivos: obtener la mayor cantidad del analito en los extractos, es decir, porcentajes de recuperación superiores al 80% y obtener extractos libres de interferencias.
Muestras frescas de 300g de cada una de las especies vegetales estudiadas (cebollín y cilantro), se cortaron en trozos pequeños y se llevaron a un procesador de alimentos eléctrico por 2 min, hasta homogeneizarlas. Entre 2,0 y 4,0g de esta preparación se colocaron en un matraz erlenmeryer de 25 mL, limpio y se adicionó 1 mL de solución metanólica conteniendo los OPs (1-40 µg.mL-1), inmediatamente, se adicionaron NaCl (0,2 g), sulfato de sodio anhidro (5 g), acetato de etilo/acetona (9 mL, 90:10) y se realizó la extracción magnética durante 15 min. Se obtuvieron extractos muy coloreados, después de la extracción preliminar, lo que requirió optimizar la limpieza de los extractos antes del análisis cromatográfico. Se realizaron ensayos de limpieza usando como fases sólidas carbón grafitado, carbón activado y mezclas en diferentes proporciones de carbón activado/florisil.
Extractos de 10mL se pasaron, en un sistema al vacío (5 mL.min-1), a través de cartuchos de 100 mg de carbón grafitado acondicionados con metanol (10 mL) y acetato de etilo/acetona (10 mL, 90:10), seguidamente, se hizo la elución de los OPs retenidos en el cartucho agregando acetato de etilo/acetona (3x 2 mL, 90:10). Estos extractos se transfirieron a un tubo de ensayo y se evaporaron bajo corriente de N2 en un baño de María (35ºC) hasta un volumen final (2 mL). Posteriormente, los extractos se transfirieron a un vial al que se le adicionó el estándar interno (TPP) y se inyectó (1,0 µL) por duplicado en el cromatógrafo de gas.
Se evaluó la eficiencia de la extracción con carbón grafitado (250 mg) y carbón activado (250 mg), manteniendo las condiciones de acondicionamiento y elución. Se realizaron modificaciones al procedimiento anteriormente descrito mezclando carbón grafitado/florisil y carbón activado/florisil (250mg:1400mg).
La eficiencia del empaque carbón activado/florisil se evaluó usando dos formas de empaque: carbón activado: florisil: carbón activado 100mg:1400mg:100mg y 150 mg:1400mg:150mg. El cartucho se acondicionó con metanol (10 mL) y acetato de etilo/acetona (10 mL, 90:10). La elución se llevó a cabo agregando la mezcla acetato de etilo/cetona (2x5 mL, 90:10).
La complejidad de los extractos y la necesidad de disminuir los costos del análisis hizo necesario llevar a cabo ensayos de limpieza adicionales, tal como el empacado del cartucho con carbón activado/florisil en menores proporciones (200:700 mg), que permitieron la reutilización de los cartuchos. La elución se realizó con acetato de etilo/acetona (7x5 mL, 90:10).
Los resultados de la optimización de los métodos de extracción y limpieza correspondientes a cada insecticida se analizaron con estadística descriptiva (media, desviación estándar, desviación estándar relativa, coeficientes de correlación de las curvas de calibración) y se realizaron análisis de varianza y pruebas de significancia (Prueba de Tukey) para comparar los métodos de extracción.
Resultados y discusión
Optimización de la separación cromatográfica
Las condiciones instrumentales del cromatógrafo de gases (Buscema et al., 2004; Ettiene et al., 1997; Prieto et al., 1999; Prieto et al., 2002), permitieron la cuantificación de los OPs en las matrices estudiadas. El tiempo para completar la separación fue de 35 min.
El cuadro 1 muestra los tiempos de retención y los factores de respuesta relativos a trifenilfosfato (TPP).
Cuadro 1. Tiempos de retención de los insecticidas organofosforados y factores de respuesta relativos a trifenilfosfato empleados para la cuantificación.
| Insecticida organofosforado | Tiempo de retención (Min) | Factor de respuesta relativo |
| Diazinon | 14,68 | 0,6971 |
| Disulfotón | 15,67 | 0,7292 |
| Fenclorfos | 17,78 | 0,6199 |
| Fenitrotión | 18,39 | 0,5969 |
| Malatión | 18,88 | 0,5049 |
| Paration | 19,52 | 0,7694 |
| Clorfenvinfos | 21,59 | 0,4505 |
| Methidatión | 22,48 | 0,4746 |
| Trifenilfosfato | 28,90 | 1,0000 |
La figura 1 muestra un cromatograma típico de la separación cromatográfica de los OPs (diazinon, disulfotón, fenclorfos, fenitrotión, malatión, paratión, clorfenvinfos, metidatión) y del TPP. El orden de elución de los compuestos fue similar a los reportados en la literatura (Holland et al., 1994). Los coeficientes de correlación lineal resultaron superiores a 0,999 para todos los OPs estudiados, indicando una alta linealidad en los intervalos de concentración estudiados.

El cuadro 2 muestra los límites de detección obtenidos para los OPs. Puede observarse que la concentración más baja correspondió a malatión (0,00168 µg.g-1). Sánchez et al., (2005) publicaron un límite de detección mayor para malatión (0,017 µg.g-1) en frutos de guayabo. Calonge (2002), señalaron mayores límites para malatión (0,010 µg.g-1) en frutos y vegetales, pero similares para fenitrotión (0,025 µg.g-1) y paratión (0,020 µg.g-1).
Cuadro 2. Límites de detección para los insecticidas en estudio.
| Insecticida organofosforado | Límites del detección (mg.g-1) |
| Diazinon | 0,006090 |
| Disulfotón | 0,005602 |
| Fenclorfos | 0,011413 |
| Fenitrotión | 0,012558 |
| Malatión | 0,001688 |
| Paratión | 0,020463 |
| Clorfenvinfos | 0,018977 |
| Metidatión | 0,012006 |
Optimización del Procedimiento de Extracción y Limpieza
Los porcentajes de recuperación obtenidos en cebollín y cilantro empleando 100 mg de carbón grafitado se presentan en la figura 2. Las muestras de cebollín presentaron bajos porcentajes de recuperación. En contraste, en cilantro se obtuvieron porcentajes de recuperación superiores. Sin embargo, a excepción de malation y clorfenvinfos, estos valores no están dentro de los intervalos de seguridad establecidos por el Departamento de Agricultura de los Estados Unidos (USDA, 1991). Por otra parte, con esta cantidad de fase (100 mg de carbón grafitado) los extractos no se limpiaron.

En función de estos resultados se planteó la necesidad de aumentar la cantidad de carbón grafitado (250 mg) y disminuir la cantidad de muestra (2,0 g). Los resultados obtenidos en cebollín, no arrojaron diferencias significativas (P>0,05) en relación con 100mg de carbón grafitado (figura 3). La recuperación fue baja para todos los OPs.
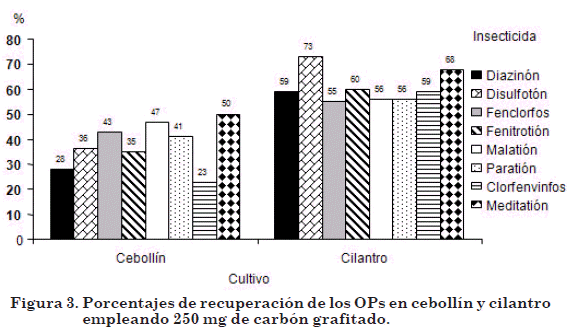
En cilantro se obtuvieron diferencias significativas (P<0,05) para disulfotón (73%). Estos bajos porcentajes de recuperación pueden deberse a la retención de los analitos en la fase sólida (Lucio et al., 2004; Prieto et al., 2002). Por otra parte, el color original de los extractos disminuyó levemente.
Estudios realizados han demostrado la eficiencia de 250 mg de carbón grafitado como fase sólida de extracción y limpieza. Así, altos porcentajes de recuperación fueron obtenidos por Ettiene et al., (1997), para malation (100%) y paratión (102%) en muestras de mosto de uvas; Prieto et al., (2002) para diazinon y malatión (75-105%), en muestras de tomate; Norman y Panton (2001) para paratión (110%), en muestras de maíz; Danis et al., (2002) para paratión (112%) en durazno fresco.
Basados en los resultados anteriores, se sustituyó el carbón grafitado por carbón activado (250 mg), lo que permitió obtener porcentajes de recuperación más altos en ambos tipos de materiales vegetales.
Se puede observar que la mayoría de los OPs se sobrerecuperó (figura 4). Esto puede deberse a coextractos que eluyen con el analito y producen un incremento en su señal cromatográfica (Prieto et al., 2002). Adicionalmente, se obtuvo una limpieza parcial de los extractos, que se evidenció por la coloración amarilla de los mismos.

En función de los resultados obtenidos con carbón grafitado y activado (250 mg), se modificó el empaque empleando cartuchos con carbón grafitado/florisil (250:1400mg) y carbón activado/florisil (250:1400mg). En las figuras 5 y 6 se muestran los porcentajes de recuperación obtenidos, los cuales fueron superiores cuando se empleó carbón activado/florisil en ambas matrices. Sin embargo, la recuperación en cilantro, resultó inferior a la obtenida en cebollín.


Los bajos porcentajes de recuperación con carbón grafitado/florisil se atribuyen a la alta afinidad de los insecticidas por esta fase (Lentza-Rizos et al., 2001; Lehotay et al., 2005; Lucio et al., 2004; Prieto et al., 2002), que se pudo comprobar cuando se ensayó con carbón grafitado puro.
En la búsqueda de la máxima eficiencia en la recuperación de todos los OPs se evaluó una nueva modalidad de empaque rellenando el cartucho en tres capas: carbón grafitado/florisil/carbón grafitado (100:1400:100mg) y carbón activado/florisil/carbón activado (100:1400:100mg).
En las figuras 7 y 8 se aprecia la misma tendencia observada con 250 mg de carbón, es decir, se obtuvieron porcentajes superiores cuando se empleó carbón activado/florisil/carbón activado (100:1400:100mg) en ambas matrices, también con las mayores recuperaciones en cebollín.


Debido a que los procedimientos ensayados no arrojaron óptimos porcentajes de recuperación para todos los OPs y en función de que los porcentajes de recuperación son superiores con carbón activado se empacó un cartucho conteniendo carbón activado/florisil/carbón activado en la proporción 150:1400:150mg. Los resultados se muestran en figura 9. Se observan tanto en cebollín como en cilantro porcentajes de recuperación superiores al 60%. Sin embargo, con esta modalidad de empaque se obtuvo un efecto memoria de fenclorfos, fenitrotión, malatión, clorfenvinfos y metidatión en cebollín, y de diazinon, disulfotón, fenclorfos, fenitrotión, paratión, clorfenvinfos y metidatión en cilantro, indicando una fuerte retención de estos OPs en la fase sólida (Lentza-Rizos et al., 2001; Lehotay et al., 2005; Lucio et al., 2004; Prieto et al., 2002; Schenck y Lehotay, 2000; Yi et al., 2006).

Para corroborar el efecto memoria, se disminuyó la cantidad de carbón activado y de florisil (200mg:700mg). En la figura 10 se observa una disminución de los porcentajes de recuperación en ambas matrices.

Con estas condiciones de limpieza se mantuvo el efecto memoria. En una segunda elución se lograron recuperar del extracto de cebollín cantidades remanentes de diazinon, disulfotón, fenitrotión y paratión, mientras que del extracto de cilantro se recuperaron diazinon, disulfotón fenclorfos, fenitrotión, paratión y clorfenvinfos, demostrándose una fuerte afinidad de estos OPs por la fase sólida.
Finalmente, se aumentó el volumen de solvente de elución (7x5 mL) para lograr la total remoción de los OPs de la fase adsorbente. La figura 11 muestra la eficiencia en la extracción de los OPs en ambas matrices.

Las desviaciones estándar relativas entre corridas (repetibilidad), resultaron menores a 3% y entre muestras (reproducibilidad) menores a 7%, indicando una alta precisión del método optimizado, por lo que se considera adecuado para su aplicación en el análisis de residuos de insecticidas OPs en cebollín y cilantro.
Conclusiones
El procedimiento de extracción y limpieza en fase sólida empleando un empaque de carbón activado/florisil (200:700mg), permitió cumplir con los dos objetivos de la etapa de extracción y limpieza, ya que se obtuvo óptimos porcentajes de recuperación (96-105%) para todos los OPs en las dos matrices y extractos libres de pigmentos.
Agradecimientos
Los autores desean expresar su agradecimiento al CONDES por el financiamiento del proyecto CC-0257-02.
Literatura citada
1. Buscema, I., G. Ettiene, D. Medina y A. Prieto. 2004. Método de extracción en fase sólida de residuos de insecticidas organofosforados en aguas naturales. Rev. Fac. Agron. (LUZ). 21:280-289. [ Links ]
2. Calonge, P. 2002. Determination of pesticides residues in fruits and vegetables by gas chromatography and NPD detector/MS. J. Chromatogr. A. 1005:215-228. [ Links ]
3. Danis, T., V. Sakkas, T. Stratis and A. Albanis. 2002. Pesticides multiresidues analysis in fresh and canned peaches using solid phase extraction and gas chromatography techniques. Bull. Environ. Contam. Toxicol. 69:674-681. [ Links ]
4. Ettiene, G., A. Prieto, D. Medina, I. Buscema, L. Sandoval y L. Unda. 1997. Residuos de insecticidas organofosforados en mosto y vino de uvas. Rev. Fac. Ing. LUZ. 20(3):223-230. [ Links ]
5. Holland, P., D. Mcnaughton y C. Malcom. 1994. Multiresidue analisys of pesticides in wines by solid-phase extraction. JAOAC. 17(1):79-86. [ Links ]
6. Itoh, H., S. Kawasaki y J. Tadano. 1996. Application of HPLC with mass spectrometry detection for pesticides analysis. J. Chromatogr. A. 754:61-76. [ Links ]
7. Kadennczki, L., Z. Arpad, I. Gardi, A. Ambrus, L. Gyorfi, G. Reese y W. Ebing. 1992. Column extraction of quantity of pesticides residues from fruit and vegetables: simple analysis method. J. Assoc. Off. Anal. Chem. 75:53-61. [ Links ]
8. Lentza-Rizos, Ch., E. Avramides y F. Cherasco. 2001. Low-temperature clean-up method for the determination of organophosphorus insecticides in olive oil. J. Chromatogr. A. 912:135-142. [ Links ]
9. Lehotay, S., A. de Kok, M. Hiemstra and P. Bodegraven. 2005. Validaton of a fast and easy method for the determination of residues from 229 pesticides in fruits and vegetables using gas and liquid chromatography and mass spectrometry detection. J. AOAC Int. 88(2):595-614. [ Links ]
10. Lucio F., C. Melo y I. Jardin. 2004. New materials for solid phase extraction and multiclass high-performance liquid chromatographic analysis of pesticides. J. of Chromatogr. A. 1032(1-2):51-58. [ Links ]
11. Miller-Miller. 1993. Estadística para la química analítica. McGraw Hill.3ra edición. México. [ Links ]
12. Mol, H., R. Van Dam y O. Steijger. 2003. Analysing residues of pesticides in plants by gas chomatography. J. Chromatogr. A. 1015:119-127. [ Links ]
13. Norman, K. and S. Panton. 2001. Supercritical fluid extraction and quantitative determination of organophosphorus pesticides in wheat and maize using gas chromatography with flame photometric and mass spectrometric detection. J. Chromatogr. A. 907:247-255. [ Links ]
14. Oliva, J., A. Barba, N. Vela, F. Melendreras y S. Navarro. 2000. Determination of chlorpiriphos, penconazol, phenarimol, vochlozolin, y methalaxil in grapes, most and wine. J. Chromatogr. A. 882:213-220. [ Links ]
15. Picó, J., y Y. Font. 2003. Determinación de residuos de plaguicidas en muestras de alimentos. Rev Toxicol. 20:166-175. [ Links ]
16. Prieto, A., G. Ettiene, D. Medina, I. Buscema, G. González and L. Araujo. 1999. Analysing organophosphorus pesticides in wines using graphitized carbon black extraction cartridges. J. Food Add. and Contam. 16(2):57-61. [ Links ]
17. Prieto, A., D. Molero, G. González, I. Buscema, G. Ettiene y D. Medina. 2002. Persistence of methamidophos, diazinon and malathion in tomatoes. Bull. Envirom. Contam. 69:479-485. [ Links ]
18. Sánchez, J., G. Ettiene, I. Buscema y D. Medina. 2005. Persistencia de malathion y clorpyriphos en guayaba. Rev. Fac. Agron. (LUZ). 22:65-75. [ Links ]
19. Schenck, F. and S. Lehotay. 2000. Does further clean-up reduce the matrix enhancement effect in gas chromatographic analysis of pesticide residues in food. J. Chromatogr. A. 868:51-61. [ Links ]
20. Stan, H.J. 2000. Analysing of residues pesticides in apples using gas chromatography with atomic emision detection. J. Chromatogr. A. 892:347. [ Links ]
21. Tang, F., Y. Yongde, H. Rimao, G. Shimei and J. Tang. 2005. Development of methods for determination of the residues of 15 pesticides in medicinal herbs Isatis indigotica Fort By capillary gas chromatography with electron capture or flame photometric detection. J. AOAC. 88 (3):720-728. [ Links ]
22. USDA. United States Department of Agriculture. 1991. Analytical Chemistry Laboratory Guidebook Residue Chemistry. Science and Technology. FSIS. Washington, D.C. [ Links ]
23. Yi, X., Q. Hua and Y. Lu. 2006. Determination of organophosphorus pesticide residues in the root of platycodon grandiflorum by solid-phase extraction and gas chromatography with flame photometric detection. J. AOAC. 89(1):225-231. [ Links ]
