











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkInvestigación Clínica
versión impresa ISSN 0535-5133
Invest. clín v.42 n.1 Maracaibo mar. 2001
Síndrome de ovarios poliquísticos de origen extraovárico. Revisión.
José Terán Dávila y Alejandro D. Teppa-Garrán.
Servicio de Salud Reproductiva, Centro Colaborador del Programa Especial de Reproducción Humana de la Organización Mundial de la Salud. Maternidad “Concepción Palacios”. Caracas, Venezuela.
Resumen.
Una esteroidogénesis ovárica anormal es uno de los hechos establecidos en el síndrome de ovarios poliquísticos (SOP). Aunque ello implica un defecto ovárico intrínseco, también puede estar influenciado por factores extraováricos. Aunque de etiología desconocida, el SOP es una de las alteraciones endocrinológicas más comunes de la práctica ginecológica. Esta entidad se manifiesta, desde hallazgos ultrasonográficos de morfología ovárica poliquística, hasta la aparición de infertilidad, hiperandrogenismo y trastornos menstruales. Existe una serie de mecanismos implicados en la elevación de los andrógenos de origen extraovárico presentes en las pacientes con SOP. Entre estos factores, están involucrados los de origen central y periférico, aunado a disfunción adrenocortical y genéticos. De esta manera, las alteraciones producidas pueden involucrar aspectos genéticos, moleculares, bioquímicos, fisiológicos y endocrinológicos, incluso, en ocasiones, varios de ellos pueden interactuar al mismo tiempo. Los niveles elevados de andrógenos séricos pueden suprimir la producción de gonadotropinas a nivel hipofisiario, ya sea directamente o como producto de su conversión periférica, además de ocasionar las manifestaciones clínicas de acné, alopecia y la detención de la maduración folicular, por lo cual estas pacientes presentan trastornos menstruales, anovulación e infertilidad. Las características del SOP de origen extraovárico incluyen la elevación de la 17-hidroxiprogesterona en respuesta al test de ACTH y la supresión de andrógenos adrenales por la dexametasona. Es posible mejorar la función ovárica de algunas pacientes con SOP, cuando se asocian los glucocorticoides al tratamiento con citrato de clomifeno para inducir la ovulación, debido a la supresión de la secreción de ACTH.
Palabras clave: Síndrome de ovarios poliquísticos, hiperandrogenismo, suprarrenal.
Extraovarian Polycystic Ovarian Syndrome. Review.
Abstract.
An established fact in the polycystic ovarian syndrome (POS) is an abnormal ovarian steroidogenesis. Though this suggest an intrinsic ovarian defect, the syndrome could also be influenced by factors outside the ovaries. Although of unknown etiology, the POS is one of the most frequent endocrine disorders in the gynecologic practice. The disorder is characterized by ultrasound findings of enlarged polycystic ovaries, hyperandrogenism, menstrual disorders, obesity and including the appearance of infertility. There are a series of mechanisms involved in the extraovarian androgen increase in patients with POS. Among these mechanisms are implicated those of central and peripheral origin, genetic factors and adrenocortical dysfunction. In the same way, the alterations produced could imply genetic, molecular biological, biochemical, physiological and endocrinological factors. Sometimes all these factors could interact at the same time. The high serum androgen level could stop the pituitary gonadotropin production, either as a direct mechanism or as a result of its peripheral conversion. The increased androgens also explain the manifestations of clinical acne, hirsutism, and the detention in follicular ovarian maturation. All these manifestations are related with the menstrual disorders, anovulation, and infertility that these patients develop. The characteristics of the extraovarian POS include the 17-hydroxyprogesterone elevation in response to the ACTH test and the dexamethasone suppression of adrenal androgens. It is possible to improve the ovarian function in some patients with POS. This could be achieved with clomiphene citrate associated with glucocorticoids to induce ovulation.
Key words: Polycystic ovarian syndrome, hyperandrogenism, adrenal gland.
Recibido: 02-05-2000. Aceptado: 18-12-2000
INTRODUCCIÓN
El síndrome de ovarios poliquísticos (SOP) es una entidad caracterizada por un desorden en la foliculogénesis asociado a una mayor tendencia en la secreción de andrógenos, que puede manifestarse con anovulación, infertilidad, oligoamenorrea, obesidad, hirsutismo, hiperinsulinemia, resistencia a la insulina y dislipidemia.1 Fundamentalmente, el origen del exceso de andrógenos es el ovario, el cual se estima estar presente en alrededor del 70% de las pacientes; no obstante, también puede presentarse un incremento de andrógenos adrenocorticales2 y, en este sentido, se ha reportado que más de la mitad de las pacientes con SOP cursan con niveles elevados de dehidroepiandrosterona sulfato (DHEAS),2,3 así como también se ha documentado la existencia de valores séricos elevados de dehidroepiandrosterona (DHEA), androstenediona (A4) y androstenediol (A5), en pacientes con SOP.4 Moltz y col.5 han realizado cateterización selectiva de venas adrenales y ováricas, y asimismo han demostrado una hipersecreción de andrógenos adrenales en pacientes con hiperandrogenismo.
Los mecanismos responsables del exceso de andrógenos extraováricos presentes en las pacientes con SOP no se conocen a plenitud; sin embargo, diversos estudios involucran la participación de factores de origen central, periférico, adrenocortical y/o genéticos, los cuales discutiremos en este capítulo, para lo cual es necesario que hagamos previamente un breve recuento sobre ciertos elementos fisiológicos.
EL EJE HIPOTÁLAMO-HIPÓFISIS-ADRENAL
Hormona hipotalámica liberadora de corticotropina (CRH)
La CRH es una hormona proteica constituida por 41 aminoácidos, que estimula la síntesis y liberación de la hormona adrenocorticotropa (ACTH).6 Es sintetizada principalmente por las motoneuronas del núcleo paraventricular del hipotálamo, y a través de la circulación portal alcanza la adenohipófisis, en donde estimula la transcripción, síntesis y secreción del gen precursor de una serie de péptidos, como la ACTH, y opioides como la b-endorfina, conocido con el nombre de proopiomelanocortina (POMC).7
La CRH posee dos patrones fisiológicos pulsátiles de secreción: uno basal, de niveles constantes y otro intermitente, como respuesta ante situaciones de estrés; además, actúa sobre la eminencia media inhibiendo la secreción de la hormona liberadora de gonadotropinas (GnRH), a través de mecanismos paracrinos y/o sinapsis axónicas entre grupos celulares.
Su mecanismo de acción molecular sobre el adrenocorticotropo, lo realiza, como cualquier hormona proteica, a través de un receptor específico en la membrana celular, que involucra como segundo mensajero al sistema adenilciclasa/monofosfato cíclico de adenosina (AC/AMPc).8
Hormona adrenocorticotropa
La ACTH es una hormona proteica constituida por 39 aminoácidos, que estimula el proceso de esteroidogénesis en la corteza de las glándulas suprarrenales y ejerce su acción sobre la regulación de la biosíntesis esteroidea de dos formas: una aguda, estimulando la secreción del cortisol como respuesta al estrés, y otra crónica, induciendo un incremento en la síntesis de RNA mensajero y de las enzimas pertenecientes a los citocromos P450scc (20,22-desmolasa), P450c11 (11b-hidroxilasa), P450c21 (21-hidroxilasa) y P450c17 (17a-hidroxilasa/17-20 desmolasa)6,9 Fig. 1.
De manera similar a la CRH, la colecistoquinina, la angiotensina II y la vasopresina estimulan la liberación de ACTH; no obstante, estos reguladores no tienen tanta efectividad como el primero.8 Además, existen otros neurotransmisores que facilitan la secreción de ACTH, como la serotonina y la acetilcolina, que estimulan directamente a ciertos receptores hipotalámicos, que responden con descargas de CRH. Sobre este particular, se puede destacar que la vasopresina, la angiotensina II, la norepinefrina y la epinefrina tienen un mecanismo de acción diferente al CRH, en lo que se refiere a la vía de segundos mensajeros utilizada en la hipófisis, ya que todas ellas utilizan como segundo mensajero al sistema calcio/calmodulina-fosfolípidos (Ca++/CaM-F).8
La secreción de CRH, ACTH y cortisol involucra patrones episódicos con variaciones diurnas, alcanzándose los máximos niveles en las primeras horas de la mañana (8 a.m.), así como un ritmo episódico o ultradiano, el cual ha sido menos estudiado8, 9 (Fig. 2).

Es importante destacar que la ACTH posee cierta actividad melanocítica, lo cual se explica porque tanto la ACTH como las hormonas estimulantes de melanocitos a y b, provienen de un gen común y, además, porque comparten algunos aminoácidos en su estructura molecular. Este hecho explica, en parte, la hiperpigmentación de la piel observada en algunas pacientes con SOP, que cursan con niveles elevados de ACTH.6
La ACTH actúa a través de receptores específicos ubicados en la membrana plasmática, utilizando el sistema adenilciclasa/AMPc para producir sus principales funciones en la glándula suprarrenal, a saber: regular la síntesis de glucocorticoides en la zona fasciculada de la corteza y la síntesis de andrógenos en la capa reticular de la misma.8 Por otra parte, es necesario recordar que Hornsby y Gill,10 han señalado el papel imprescindible del ion calcio para que tenga lugar la unión entre la ACTH y su receptor.
Los glucocorticoides
Son hormonas esteroides de 21 átomos de carbono, pertenecientes al grupo ciclopentanoperhidrofenantreno, con grupos hidroxilos en las posiciones 11, 17 y 21, un grupo cetónico en la posición 3 y un doble enlace en la posición 4,511 (Fig. 3).
El cortisol es el esteroide natural más importante del cuerpo humano y el principal glucocorticoide, ya que posee el 95% de esta actividad en toda la aconomía. El paso limitante en la síntesis del cortisol es la conversión del colesterol en pregnenolona (P5), paso que es regulado por la ACTH, quien estimula el clivaje enzimático de la cadena lateral del carbono 20 realizado por la 20,22 desmolasa o liasa. El cortisol circula en el plasma unido en un 75% a una proteína a2-globulina llamada transcortina, y el resto unido a la albúmina o en forma libre. Los corticosteroides son metabolizados principalmente en el hígado, donde son conjugados en forma de glucorónidos, y en poca cantidad como sulfatos, y son excretados alrededor del 75% por la orina y el resto por las heces.6

Los andrógenos
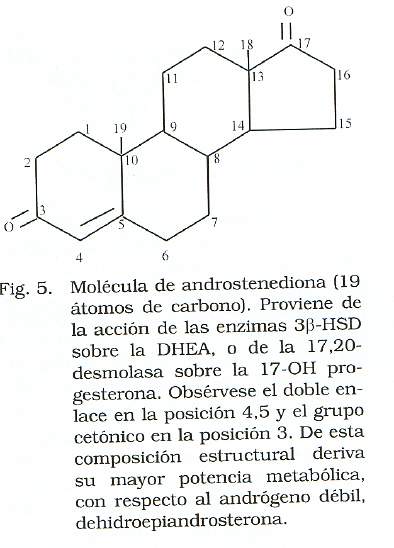
Los andrógenos son moléculas que provienen del colesterol y están conformados por 19 átomos de carbono. De acuerdo a su composición estructural deriva la potencia metabólica; es decir, si presentan un doble enlace en la posición 5,6 y un grupo hidroxilo en el carbono 3, son andrógenos débiles, mientras que los de mayor potencia se caracterizan por poseer un grupo cetónico en la posición 3 y el doble enlace en la posición 4,5 (Figs. 4 y 5).
Como principales andrógenos se pueden señalar: la testosterona, la dehidrotestosterona, la A4 y la DHEA y su metabolito sulfatado, DHEAS,12 los cuales son secretados por la corteza adrenal y los ovarios y, por otra parte, derivan de la conversión de ciertos esteroides en el hígado y tejidos periféricos (tejido adiposo, músculo, piel, etc.). La A4 ejerce una potencia androgénica equivalente a sólo el 10% al 20% de la testosterona, de acuerdo a determinaciones realizadas mediante bioensayos, mientras la DHEA posee solamente el 5% de esa potencia, aunque el andrógeno más potente es la dehidrotestosterona, con una potencia de 1,5 a 3 veces superior a la testosterona, la cual proviene de la acción de la 5a-reductasa sobre la testosterona. Del total de testosterona, el 50% proviene de la conversión periférica de la A4, mientras el resto es producido en partes iguales por los ovarios y las glándulas adrenales. La A4 es producida en cantidades similares por los ovarios y la corteza adrenal; sin embargo, el 90% de la DHEA y el 99% de la DHEAS son producidos por la corteza adrenal12 (Tabla I).
Estas hormonas son importantes en las mujeres para la estimulación del crecimiento del vello corporal, la fuerza muscular, el balance positivo de nitrógeno y la libido, entre otros. En concentraciones anormalmente elevadas, como en el SOP, pueden ejercer efectos masculinizantes, tales como: seborrea, calvicie, trastornos menstruales, clitoromegalia, engrosamiento del tono de la voz, desarrollo muscular, etc. Por otra parte, la DHEA, DHEAS y la A4 presentan un ritmo circadiano semejante al del cortisol, es decir, exhiben un cenit matutino y un nadir vespertino.12

La corteza adrenal
La glándula suprarrenal contiene dos órganos endocrinos diferentes: la corteza y la médula; no obstante, sólo haremos referencia a la corteza suprarrenal por su papel involucrado en la síntesis de esteroides (Fig. 6).
FUENTE DE LOS ANDRÓGENOS EN LA MUJER ADULTA
| A4 (%) | T (%) | DHEA (%) | DHEAS (%) | DHT (%) | |
| Ovario | 49,5 % | 25% | <1,5% | <0,5% | <0,1% |
| Corteza suprarrenal | 49,5 % | 25% | 95% | >99% | <0,1% |
| Conversión periférica | 1% | 50% | <3,5% | <0,5% | >99% |
(A4) androstenediona. (T) tetosterona. (DHEA) dehidroepiandrosterona. (DHEAS) dehidroepiandrosterona sulfato. (DHT) dehidrotestosterona.

Embriológicamente, la corteza suprarrenal se desarrolla a partir del epitelio celómico de la superficie media del pliegue urogenital.6 Este mismo epitelio origina las células granulosas del ovario. La corteza adrenal ocupa alrededor del 80% al 90% del volumen de la glándula suprarrenal y está conformada por tres zonas con funciones diferentes: la zona más externa o glomerular, la cual sintetiza mineralocorticoides, principalmente aldosterona; la zona media o fascicular, que secreta glucocorticoides (cortisol); y la zona interna o reticular, que sintetiza andrógenos, fundamentalmente DHEA.6 Además, la corteza adrenal sintetiza y libera pequeñas cantidades de estrógenos, testosterona y A4.6
La corteza suprarrenal produce todos estos compuestos a partir del mismo precursor: el colesterol, y mediante diferentes complejos enzimáticos, pertenecientes al citocromo P450.6 El colesterol es sintetizado, en parte por la glándula suprarrenal, pero la mayor parte deriva de las lipoproteínas de baja densidad (LDL) circulantes.

MECANISMOS RESPONSABLES DEL EXCESO DE ANDRÓGENOS EXTRAOVÁRICOS PRESENTES
EN LAS PACIENTES CON SOP
La inmensa mayoría de las pacientes que presentan un hiperandrogenismo suprarrenal funcional tienen origen idiopático. De esta manera, cuando se establece una función suprarrenal elevada, solamente en el 10% de las pacientes es posible confirmar un elemento fisiopatológico con exactitud, como por ejemplo una hiperplasia adrenal congénita no clásica.13 No obstante, existen varios factores causales del hiperandrogenismo adrenal en las pacientes con SOP, los cuales involucran un origen central, periférico, adrenocortical y/o genético y, además, cabe señalar a la pseudociesis, que la citamos como una variante del SOP extraovárico. Todos estos factores los ampliaremos a continuación:
a) Factores centrales: disfunción eje HHA, incremento secreción o sensibilidad de la hormona estimulante de la corteza adrenal, estrés psicológico, endorfinas, catecolaminas, prolactina.
b) Factores periféricos: alteraciones en el metabolismo de los andrógenos adrenales, obesidad, estrógenos, andrógenos ováricos, insulina y leptina.
c) Disfunción adrenocortical: hiperactividad adrenocortical, hiperplasia adrenal congénita (variedad no clásica), deficiencia de la enzima 11-hidroxilasa, alteraciones en la regulación del citocromo P450c17a, deficiencia de la enzima 3b-HSD, deficiencia de la enzima 11-deshidrogenasa, estearasa de colesterol, adrenarca exagerada.
d) Factores genéticos y pseudociesis (variante del SOP).
FACTORES CENTRALES
Disfunción del eje Hipotálamo-Hipófisis-Adrenal (HHA)
La participación del eje HHA puede observarse, en parte, de acuerdo al comportamiento de la ACTH. De esta manera, podemos señalar que la regulación de ACTH, al involucrar estímulos de retroalimentación negativa ejercidos por los glucocorticoides, además de efectos estimulatorios por parte de CRH, vasopresina, angiotensina II y norepinefrina, podría involucrar alteraciones en el ámbito fisiológico, bioquímico o molecular, por lo menos conceptualmente. Sin embargo, en la práctica no se han encontrado diferencias en cuanto a niveles séricos, ritmo circadiano y pulsatilidad de la ACTH entre pacientes con SOP y controles, así como ninguna correlación entre los valores de DHEAS y de ACTH en sangre.14 En esta misma línea de investigación se encuentran Azziz y col.2 quienes no han encontrado alteraciones del eje HHA, y señalan que no existe una exagerada secreción hipofisiaria de ACTH a la estimulación por CRH, en pacientes con SOP. Por otra parte Güven y col.15 indican que no existen diferencias, tanto en el número como en la afinidad de los receptores para glucocorticoides, entre las pacientes con SOP y los controles, que pudieran sugerir alteraciones del eje HHA en estas pacientes. Aunque por otra parte, Invitti y col.16 reportaron que puede existir cierto desorden en la secreción de cortisol en las pacientes con SOP, debido a un discreto incremento en la frecuencia y amplitud de los pulsos de ACTH. Por otra parte, existe una mayor secreción de DHEA al estímulo de la ACTH, sin modificar los niveles de cortisol.12 No obstante, es posible que las alteraciones en el eje HHA descritas en algunas pacientes con SOP, sólo estén presentes en aquellas pacientes con exceso de andrógenos adrenales.
Hormona estimulante de la corteza adrenal (CASH, en inglés)
Se ha propuesto la existencia de una hormona de origen hipofisiario, que actúa sobre la corteza adrenal directamente, la cual se presume deriva de la misma molécula precursora que la ACTH y que sería responsable del exceso de andrógenos adrenales en ciertos pacientes con SOP.17 Esta hormona, es un polipéptido regulador ectópico de la síntesis de andrógenos, el cual mantiene cierta relación con la ACTH. En el caso particular del SOP, la relación ACTH/CASH se encuentra disminuida, lo cual favorece la producción de andrógenos; mientras que si la relación está aumentada, hay tendencia a mayor producción de cortisol.18
Estrés psicológico, endorfinas y catecolaminas
Orenstein y col.19 han reportado que las pacientes con hiperandrogenismo presentan usualmente algún grado de sintomatología psicosexual, depresión o ansiedad. Por tanto, el estrés psicológico puede afectar al eje HHA, cuyo mecanismo podría explicarse por alteraciones en la secreción de ACTH u otros productos de la POMC, incluyendo a las endorfinas, así como por la descarga de catecolaminas por el sistema nervioso central (SNC) o la médula adrenal, o una mayor sensibilidad adrenocortical.12 Cualquier situación de estrés puede incrementar hasta 6 veces los niveles circadianos de CRH, asociado a su vez con elevaciones de los niveles de ACTH y corticosterona. La hiperproducción de ACTH, como respuesta al estrés, involucra la estimulación del sistema límbico, fundamentalmente de la región del hipocampo y la amígdala.6 Es conocido que durante una respuesta al estrés se descargan a la circulación fragmentos de POMC, tales como ACTH, lipotropina (LPH) y b-endorfina. Así mismo, Givens y col.20 reportaron niveles incrementados de b-endorfina y b-LPH en pacientes con hirsutismo, hiperandrogenismo y oligoamenorrea en comparación con los controles. Además, Lobo y col.21 señalan una significativa correlación entre valores séricos de b-endorfina inmunoreactiva y de DHEAS, tanto en pacientes con SOP como en controles. Se ha sugerido que los opioides endógenos ejercen un control inhibitorio sobre el eje HHA, el cual pudiera estar reducido o inoperativo en el caso de las pacientes con SOP con hipersecreción de andrógenos adrenales.22 En forma similar a lo sucedido con los productos del POMC, el estrés también incrementa la secreción de catecolaminas. En este sentido, Lobo y col.23 demostraron una secreción aumentada de metabolitos de las catecolaminas en pacientes con SOP, los cuales puedan estar asociados al aumento de los niveles de andrógenos adrenales circulantes.
Adicionalmente, Kleitman y Holzwarth,24 han reportado la presencia de axones de tipo noradrenérgico y dopaminérgico en la corteza suprarrenal. Existen evidencias que sugieren un importante papel en la esteroidogénesis de estos axones adrenocorticales de la corteza suprarrenal, los cuales pudieran estar comprometidos en el caso de algunas pacientes con SOP de origen extraovárico.
Hiperprolactinemia
Entre los mecanismos responsables del aumento de andrógenos adrenales es conocido la participación de la prolactina.25 Desde hace más de 20 años se han detectado receptores para prolactina en la glándula suprarrenal de animales de experimentación26 y, en otros estudios, realizados en pacientes con hiperprolactinemia, se han encontrado mayores niveles de DHEAS, testosterona, androstenediol y A4 libres.25 Es posible, que la hiperprolactinemia esté involucrada en el incremento de los niveles de DHEAS en algunas de las pacientes con SOP, posiblemente como consecuencia de una estimulación directa o, más bien, secundario a una mayor concentración de estrona (E1), producida a su vez por una mayor aromatización de la DHEAS circulante.12
Es interesante señalar, que en algunos casos de pacientes anovulatorias con SOP, que presentan concomitantemente galactorrea y/o hiperprolactinemia, tratadas infructuosamente con citrato de clomifeno, se ha podido inducir la ovulación al asociar 2-bromo-alfa-ergocriptina al tratamiento.27 Sobre este particular, Lobo y col.28 han reportado una disminución de los niveles de DHEAS concomitante a una baja similar de los valores séricos de prolactina en pacientes con hiperprolactinemia tratados con el mismo medicamento.
FACTORES PERIFÉRICOS
Alteraciones en el metabolismo de los andrógenos de origen adrenal
Un compromiso en el metabolismo de los andrógenos de origen adrenal se puede observar, a menudo, en las pacientes con SOP. Por esto, un trastorno en el aclaramiento (clearance) de DHEA o de DHEAS puede producir la elevación de estos andrógenos en estas pacientes. En este sentido, Haning y col.29 han encontrado una mayor producción y menor aclaramiento de DHEAS en pacientes con hirsutismo respecto a controles. Además, se puede asociar alteraciones en la velocidad de sulfatación y/o de desulfatación de la DHEA.12 Por otra parte, se ha especulado que la liberación pulsátil anormal de andrógenos podría alterar el patrón de secreción por pulsos de la hormona luteinizante (LH).30
La obesidad
Cabe destacar que la mayoría de las pacientes con SOP presentan algún grado de obesidad; por tanto, es posible pensar que esta entidad tenga alguna participación en el origen del hiperandrogenismo. El SOP es una condición familiar, cuyo origen probablemente se remonta a la adolescencia, y se relaciona con aumento de la ganancia de peso durante la pubertad.31 Los mecanismos por los cuales se desarrolla el SOP en las pacientes obesas son diferentes a los presentes en las mujeres delgadas. En general, las mujeres obesas se caracterizan por presentar un aumento de la resistencia periférica a la insulina, mientras que las mujeres delgadas con SOP, el desorden básico consiste en aumento de la pulsatilidad de LH y de la hormona del crecimiento (GH).32
Las mujeres obesas presentan un mayor grado de lipólisis en el tejido adiposo abdominal, lo cual incrementa el nivel de ácidos grasos en la circulación portal y, como consecuencia, se dificulta la extracción hepática de insulina y de este modo se inicia, o se empeora, el grado de hiperinsulinismo.33 Por otra parte, el aclaramiento de los andrógenos adrenales parece que es mayor en las mujeres obesas, lo cual se explica, principalmente, por la mayor aromatización periférica existente y por un bajo grado de actividad de la enzima 17b-hidroxiesteroide deshidrogenasa (17b-HSD) que promueve la conversión de A4 a testosterona y, además, por la acumulación de lípidos en los adipocitos y trastornos en la tasa de conjugación de las hormonas esteroideas.12
Los estrógenos
La conversión periférica de andrógenos a estrógenos puede suprimir la producción de gonadotropinas a nivel hipofisiario, debido a la retroalimentación negativa ejercida por la E1, lo cual puede conducir a anovulación crónica.34 Por otra parte, la mayor cantidad de DHEAS circulante, produce grandes cantidades de E1 al ser aromatizado,12 lo cual puede causar, a su vez, hiperprolactinemia.
Andrógenos de origen ovárico
Se ha señalado como causa de la disfunción adrenocortical en pacientes con SOP una secreción aumentada de andrógenos de origen ovárico. Se ha encontrado que las pacientes con niveles elevados de testosterona presentan a su vez mayores valores séricos de 17a-hidroxiprogesterona (17 a-OH-P4) y una mayor relación 17a-OH-P4/cortisol posterior a una prueba de estimulación adrenal.12 En un intento por estudiar el papel de los andrógenos ováricos en la función adrenocortical, Steingold y col.35 suprimieron la secreción de gonadotropinas y esteroides ováricos mediante la administración de agonistas de GnRH; no obstante, aunque el tratamiento normalizó los niveles de A4 y de testosterona, el cambio en los valores de DHEAS no fue significativo. Por tanto, los andrógenos de origen ovárico parecen tener poco impacto sobre la esteroidogénesis adrenocortical y los niveles de andrógenos adrenales circulantes.
La hiperandrogenemia es responsable de las manifestaciones pilosebáceas del síndrome, así como de la detención de la maduración folicular en los ovarios y, en consecuencia, de que los folículos presenten atresia antes de la aparición del folículo dominante, lo cual establece el mecanismo por el cual se produce la anovulación.
Hiperinsulinemia
Existen evidencias que señalan que el hiperinsulinismo interfiere con la regulación adrenal de la esteroidogénesis, permitiendo una secreción excesiva de andrógenos adrenales en respuesta a la ACTH. Esta elevación de los niveles séricos de insulina se relaciona con una menor síntesis hepática de las proteínas que unen y transportan a los factores de crecimiento insulínico (IGFBP, por sus siglas en inglés) y de la globulina transportadora de hormonas sexuales (SHBG, por sus siglas en inglés), las cuales modulan la biodisponibilidad de los factores de crecimiento insulínico y de los esteroides sexuales respectivamente.32 Por otra parte, existen estudios que demuestran una correlación inversa entre los valores séricos de insulina y de DHEAS.12 Algunas pacientes con SOP presentan una unión normal de la insulina a su receptor, pero presentan una disminución de los eventos de activación posterior. Esta resistencia a la insulina ocurre, en su mayoría, en el tejido adiposo y en los músculos, y como consecuencia, existe mayor secreción de insulina para mantener niveles normales de glucosa y, además, algunas mujeres con SOP tienen un defecto relativo en la secreción de insulina.36 Auchus y col,37 han propuesto la hipótesis que una enzima de tipo quinasa serina/treonina sobreactivada, hiperfosforila tanto el receptor de insulina como el P450c17 en mujeres con SOP, ocasionando las manifestaciones de resistencia a la insulina y el hiperandrogenismo característicos de la enfermedad.
Hiperleptinemia
La leptina es una hormona producida principalmente por el tejido adiposo, que afecta el apetito, y sus concentraciones séricas se correlacionan significativamente con la cantidad de tejido graso corporal. A nivel hipotalámico inhibe el neuropéptido Y, el cual interfiere, a su vez, con la pulsatilidad de la GnRH. Con relación a esta hormona, Vicennati y col.38 encontraron que los niveles de ésta en las mujeres obesas con SOP, son mayores que los observados en mujeres controles obesas y normales. Este hallazgo reviste interés, pues existen trabajos que relacionan la hiperleptinemia con la resistencia a la insulina.39
DISFUNCIÓN ADRENOCORTICAL
Hiperactividad adrenocortical
Es posible que el exceso en la producción de andrógenos adrenales sea consecuencia de una hiperactividad adrenocortical. En este sentido, podemos citar el trabajo de Gross y col.40 quienes reportaron una mayor captación de iodocolesterol por la glándula suprarrenal en pacientes con SOP comparado con mujeres normales. Como los niveles de ACTH no se modifican en la mayoría de las pacientes con SOP, es posible que la hiperactividad adrenocortical esté dada por una mayor sensibilidad de la zona reticular de la corteza suprarrenal a la acción de la ACTH o del sistema enzimático de clivaje del colesterol.12
Azziz y col.2 ha señalado que no existe una mayor sensibilidad de la corteza adrenal a la ACTH, sino más bien, una mayor respuesta de la corteza adrenal a la secreción de DHEAS y A4. De lo anterior, estos autores concluyen que, en estas pacientes existe una disfunción intrínseca de la corteza adrenal y no una respuesta alterada del control que ejerce el eje HHA.2 No obstante aclaran que, aún se desconoce si la causa de la disfunción intrínseca adrenal se debe a un incremento de la masa reticular o a una actividad diferente de la P450c17a.2 Además, Erel y col.41 han indicado que la respuesta androgénica adrenal a la estimulación por ACTH es normal en pacientes con SOP. No obstante, es oportuno recordar que existe un considerable número de hormonas y factores de crecimiento que participan en la esteroidogénesis en respuesta a la ACTH, los cuales podrían estar involucrados en la hiperactividad adrenocortical. En resumen, por todo lo anterior, parece existir una adaptación a la reactividad adrenal por la ACTH endógena, en pacientes con SOP.42
Hiperplasia adrenal congénita (HAC)
El SOP es una entidad que puede tener su origen en un trastorno funcional de la glándula suprarrenal, el cual puede deberse, con relativa frecuencia, a un trastorno genético conocido como HAC. Ahora bien, entre las deficiencias enzimáticas adrenales que causan la HAC, es importante destacar aquellas relacionadas con la hiperandrogenemia de las pacientes con SOP, entre las cuales destacan: 1- las deficiencias de las enzimas 11b y 21 hidroxilasa (que cursan con niveles elevados de andrógenos de la serie delta 4), 2- la deficiencia de la 3b-HSD (que presentan una producción aumentada de andrógenos de la serie delta 5) y 3- la deficiencia de la 11b-deshidrogenasa (donde ocurre mayor oxidación de cortisol a cortisona).43
Como ya hemos visto, no todos los hiperandrogenismos de origen adrenal se deben a deficiencia enzimática; sin embargo, cuando ella ocurre el más común es la deficiencia de la 21-hidroxilasa. Sabemos que la HAC se debe en el 90% de los casos a una deficiencia de la enzima antes señalada, y de acuerdo al grado de bloqueo de la misma ocurren las manifestaciones clínicas. En líneas generales, este trastorno está presente desde la séptima semana de vida intrauterina, momento en el cual comienza a funcionar la glándula suprarrenal fetal. En el caso de la deficiencia de la enzima 21-hidroxilasa, obviamente la corteza adrenal fetal no produce las cantidades de cortisol necesarias y, por tanto, se establece una retroalimentación a nivel hipotalámico para sintetizar y liberar más CRH, el cual producirá a su vez más ACTH, y esta última activará al sistema enzimático de la 20,22-desmolasa (el cual forma P5 a partir del colesterol). De los hechos anteriores, se establece un ciclo tendiente a mantener niveles normales de cortisol, pero a expensas de una hiperplasia de la glándula y, por ende, aumentan las cantidades de las hormonas precursoras al bloqueo, particularmente las de la 17a-OH-P4 y de P4, las cuales se pueden medir en el laboratorio, mediante pruebas de radioinmunoensayo o de enzimoinmunoensayo. Desde luego, que al existir una elevación de esta hormona, tambien ocurre un incremento de los valores séricos de A4, junto a la testosterona y DHEA, debido a que al incrementarse los valores de 17a-OH-P4, se desvía la ruta metabólica, lo cual es facilitado por la enzima 17,20 desmolasa, que produce clivaje entre los carbonos 17 y 20, formando esteroides de 19 átomos de carbono (A4), que son convertidos en testosterona por la acción de la 17b-HSD; todo lo cual condiciona un hiperandrogenismo y, finalmente, se establece un SOP de origen extraovárico.
La HAC, ocurre por un defecto congénito en el cromosoma 6, de herencia autosómica recesiva. Existe la forma clásica, que se presenta en el recién nacido, quienes presentan muchas veces alteraciones de los genitales externos (Fig. 7). La otra forma es la no clásica, la cual se manifiesta en el adulto, observándose generalmente después de la pubertad, la cual se relaciona con el SOP de origen extraovárico. Además, puede haber una variedad perdedora de sal cuando el bloqueo de la 21-hidroxilasa es mayor, y se encuentra comprometida la vía que conduce a la síntesis de mineralocorticoides. Las manifestaciones clínicas de la HAC en su variedad no clásica incluyen: acné (Fig. 8), hirsutismo (Fig. 9), alopecia, calvicie, trastornos menstruales e infertilidad.

Fig. 7. Paciente del Servicio de Salud Reproductiva de la Mmaternidad Concepción Palacios. Notese la fusión de los pliegues labio-escrotales, el aspecto escrotalizado de los labios mayores y la redundancia del capuchón del clítoris.
Deficiencia de la enzima 11b-hidroxilasa
Esta alteración es producida por falla en la regulación de un gen situado en el brazo corto del cromosoma 8. Esta enzima es sintetizada en la mitocondria y cataliza la transformación a corticosterona y cortisol, a partir de los sustratos respectivos desoxicorticosterona (DOCA) y 11-desoxicortisol, los cuales se acumularán, por causa del bloqueo, además de P4 y 17a-OH-P4. Como cabe esperar, disminuirá la producción de cortisol y la vía de formación de los productos del grupo pregnano se desviará hacia el grupo androstano. Hay que destacar que puede manifestarse hipertensión arterial, como un síntoma adicional, debido al efecto mineralocorticoide de la DOCA.44
Alteraciones en la regulación del citocromo P450c17a
Se han propuesto alteraciones en la regulación del citocromo P450c17a como causas de la hiperandrogenemia de origen ovárico en pacientes con SOP (gen CYP17).45,46 Este complejo enzimático presenta actividad de 17-hidroxilasa y 17,20-desmolasa, la cual está presente tanto en el tejido ovárico como en el adrenal. El citocromo P450c17a media las reacciones correspondientes a las 17 hidroxilaciones de la P5 y de la P4 y en las rupturas posteriores del puente de unión que existe entre los carbonos 17 y 20 de la 17a-OH-P5 y de la 17a-OH-P4, dando lugar a la formación de DHEA y A4, respectivamente.9 La deficiencia del citocromo P450c17a ocasiona una disminución en la síntesis de esteroides sexuales y de los glucocorticoides, en tanto que aumentan los mineralocorticoides.

Fig. 8. Paciente con bloqueo parcial de la enzima 21-hidroxilasa del Servicio de Salud Reproductiva de la Maternidad Concepción Palacios. Nótese la presencia del acné.
Se ha postulado que el mecanismo por el cual coexisten frecuentemente el hiperandrogenismo adrenal y ovárico en mujeres con SOP, resulta de una disfunción envolviendo al citocromo P450c17a en ambas glándulas. Sin embargo, la secreción adrenal anormal es particularmente prominente en la vía delta 5, mientras en el ovario es la vía delta 4. Se ha propuesto a la hiperinsulinemia como la principal causa de tal alteración.47
Deficiencia de la enzima 3b-HSD adrenocortical
También, se ha descrito en pacientes con hiperandrogenemia cierta deficiencia de la actividad de la enzima 3 b-HSD adrenocortical, la cual podría ser consecuencia de la acción de factores extraadrenales.48 La 3b-HSD es una enzima microsomal del retículo endoplásmico que circunda a la mitocondria, presente en la glándula suprarrenal, el ovario, y en tejidos periféricos como el testículo, el hígado y el riñón; la cual convierte los esteroides de la serie delta 5, derivados del colesterol, a la serie delta 4 (de gran actividad esteroidea).9 La deficiencia severa de esta enzima, en las glándulas suprarrenales y en las gónadas, es una causa conocida de hiperplasia adrenal congénita (HAC); no obstante, la deficiencia parcial de 3b-HSD se ha relacionado con hiperandrogenismo, trastornos menstruales e hirsutismo. Es fundamental esta enzima, pues su deficiencia afecta la síntesis de los esteroides sexuales, los glucocorticoides y mineralocorticoides, de esta manera, se elevarán los niveles de P5 y DHEA, aumentando la relación delta 5/delta 4, sin variación del valor de 17a-OH-P4 .43

Fig. 9. Paciente con bloqueo parcial de la enzima 21-hidroxilasa del Servicio de Salud Reproductiva de la Maternidad Concepción Palacios. Nótese la presencia del hirsutismo.
Existen dos isoformas de la enzima 3 b-HSD, las cuales son codificadas por diferentes genes y con pesos moleculares distintos. El gen 3b-HSD tipo I, expresado en el sincitiotrofoblasto y algunos tejidos periféricos, y el gen 3b-HSD tipo II, que se expresa en la capa fascicular de la glándula suprarrenal y en las gónadas.9 Sin embargo, estudios recientes señalan que sólo la deficiencia severa de 3b-HSD es producida por mutaciones en el gen 3b-HSD tipo II; por tanto, la deficiencia parcial de 3b-HSD no es una variedad de HAC.49
Deficiencia de la enzima 11 deshidrogenasa
Esta enzima convierte el cortisol en corticosterona, la cual presenta menos potencia glucocorticoide que el cortisol y, por tanto, la retroalimentación negativa sobre el hipotálamo será menor, a lo que éste responderá aumentando CRH, con igual respuesta de ACTH, y mayor estímulo sobre la glándula suprarrenal.43
La enzima estearasa de colesterol
Esta enzima libera de los cúmulos de colesterol a la fracción libre que será el sustrato para la síntesis de las hormonas esteroideas, y es estimulada por la ACTH. Alteraciones en la síntesis y secreción de esta enzima, se ha relacionado con manifestaciones de hiperandrogenismo como: acné, alopecia, hirsutismo y trastornos menstruales.6
La adrenarca exagerada
Normalmente, alrededor de los 7 años de edad se inicia la adrenarca, la cual es un factor fisiológico importante en el inicio de la maduración del eje hipotálamo-hipófisis-ovario y, por tanto, de la pubertad, la cual está caracterizada por una mayor secreción de andrógenos de origen adrenal, de los cuales destacan la DHEA, DHEAS y A4, que también son capaces de transformarse en estrógenos en los tejidos periféricos.50
Las niñas con adrenarca prematura presentan mayor probabilidad para desarrollar SOP.37 Ésta no se corresponde con un aumento de la secreción de ACTH ni de cortisol, lo cual sugiere una mayor sensibilidad de la corteza suprarrenal a esta hormona, mediante la interacción de factores de maduración aún no conocidos.37,50 Histopatológicamente, la adrenarca se correlaciona con la aparición de una zona reticular suprarrenal continua, mientras bioquímicamente se corresponde con un aumento y disminución de la actividad del citocromo P450c17a y de 3b-HSD, respectivamente.12 Se ha planteado la hipótesis de que en algunos pacientes con SOP la causa pueda ser una adrenarca exagerada y de esta manera, la hiperandrogenemia, asociada a un hiperestrogenismo, se combinen para producir una disfunción hipotálamo-hipofisiaria en estos pacientes, caracterizada por mayores concentraciones de hormona luteinizante circulante, la cual estimularía, marcadamente, la secreción de andrógenos por la teca ovárica, y manteniendo concomitantemente el hiperandrogenismo adrenal.12
FACTORES GENÉTICOS
El SOP es una condición familiar, por lo que existen cierto número de genes implicados, aunque no estudiados en su totalidad, cuya alteración se asocia a los cambios ponderales, metabólicos y hormonales característicos del síndrome. Kahsar-Miller y Azziz51 han sugerido que el SOP es producto de una alteración de tipo autosómica dominante, que se presenta con un fenotipo variable.51 Inclusive, por esta sugerencia de herencia de tipo autosómica dominante, se ha identificado un genotipo masculino caracterizado por calvicie prematura frontoparietal de inicio antes de los 30 años de edad.52 Más específicamente, la deficiencia de la enzima 21 hidroxilasa (catalizada por el citocromo P450c21), resulta en la acumulación de los precursores enzimáticos, incluyendo a los andrógenos adrenales, necesarios para la síntesis de aldosterona y/o cortisol. Este defecto, expresa una alteración del gen identificado como CYP21B, el cual se encuentra ubicado en el centro del locus del antígeno leucocitario humano.12,44
Ciertas investigaciones se han concentrado en la posible función del CYP17 (el gen que codifica a la P450C17a), debido a los estudios clínicos que señalan una regulación anormal del complejo enzimático 17-hidroxilasa/17,20 desmolasa, el cual es un conocido paso limitante en la síntesis de andrógenos. Sin embargo, se han realizado estudios en familias con SOP usando marcadores de polimorfismo próximos a este gen, con los cuales se ha excluido al CYP17 como uno de los implicados en la patología del SOP.53
De algún modo, las diferencias en la expresión del gen CYP11a, podrían explicar la variación en la producción de andrógenos en pacientes con SOP. Este gen es el productor del P450 scc (el citocromo responsable de la ruptura de la cadena lateral del colesterol).53
Por otra parte, existen estudios que indican la posibilidad de que los genes implicados en la secreción y acción de la insulina puedan tener un importante papel en la etiología del SOP. Sobre este particular, Waterworth y col.54 reportaron que en mujeres con un alelo clase III para el gen de la insulina VNTR (variable number of tandem repeats), son más obesas y presentan mayores niveles séricos de insulina, que las que tienen un alelo clase II para dicho gen.
En resumen, los estudios señalan que la herencia del SOP es de tipo oligogénica, aunque en ciertas familias las alteraciones de algunos genes son heredadas de manera dominante. De esta manera, el SOP parece ser originado por la alteración de unos pocos genes envueltos en la síntesis de esteroides, y en la acción y secreción de la insulina, lo cual se puede asociar a factores ambientales, particularmente nutricionales, todo lo cual conduce a la heterogeneidad bioquímica y clínica del SOP.52
LA PSEUDOCIESIS COMO VARIANTE DEL SOP
Esta entidad comparte algunos elementos endocrinológicos, físicos y ecosonográficos con el SOP. Sin embargo, puede tratarse de una entidad distinta, aunque persiste en la literatura mundial cierta tendencia a considerarla como una variante del SOP.
La pseudociesis es conocida también como embarazo imaginario o fantasma; es un desorden neuroendocrino, en la cual una mujer no psicótica desarrolla signos y síntomas de embarazo, tales como cambios en los órganos reproductivos, conducta materna y anovulación.55 Este trastorno psicosomático, se observa con más frecuencia en ciertas culturas, donde la mujer presenta un deseo vehemente por resultar embarazada.56 En diversas series, se ha reportado en las mujeres con pseudociesis la presencia de hiperandrogenemia, galactorrea, trastornos menstruales, infertilidad, hirsutismo, clitoromegalia (>> 1 cm) y elevación de los niveles básales de LH, testosterona y de prolactina.55 Inclusive, Bray y col,57 señalan la existencia de un hiperandrogenismo similar al presente en las pacientes con SOP.55 La prueba de TRH (con 500 µg) provoca una respuesta exagerada de LH, lo cual sugiere un trastorno dopaminérgico y, a su vez, la prueba de GnRH, ocasiona una respuesta aumentada de la LH. Sobre este particular, Terán y Febres,58 evaluaron un grupo de pacientes con pseudociesis, observando una exagerada respuesta a la LH y prolactina, mientras los niveles de FSH y de TSH fueron normales. Llama la atención en este estudio, el hallazgo, en dos de las pacientes, de una patología orgánica subyacente (hiperprolactinemia con microadenoma hipofisiario y bloqueo parcial de la 21-hidroxilasa), en la cual los síntomas se asociaron al deseo de embarazo. De esta manera, por todo lo anterior, muchos autores incluyen a la pseudociesis como una variante del síndrome de ovarios poliquísticos.55
PRUEBAS HORMONALES
Es muy importante, para tratar de orientarnos en la contribución de la glándula suprarrenal como causante del SOP, la medición de los niveles de las hormonas de origen adrenal. Es precisamente la 17a-OH-P4 la que está elevada de acuerdo al grado de bloqueo, cuando la vía hacia la formación del cortisol está interrumpida, particularmente en los casos de deficiencia de la enzima 21-hidroxilasa. Además, se miden los valores de los andrógenos DHEA y DHEAS.
También es posible hacer pruebas de supresión, como administrar dexametasona, la cual en los casos de HAC, elimina los niveles androgénicos y reduce los valores séricos de 17a-OH-P4 y DOCA. En este caso, se administra 1 mg de dexametasona, dosis que se indicará a las 11 p.m. en caso de medir la cantidad de cortisol sanguínea a las 8.00 a.m. del día siguiente.44
Es importante destacar, que cuando se presenta un SOP de origen extraovárico, si realizamos una prueba con agonistas de GnRH, la hiperandrogenemia persistirá; mientras las alteraciones serán suprimidas si el origen del SOP está en el ovario.43 Esta prueba de GnRH consiste en administrar un análogo de GnRH como la nafarelina o buserelina, a lo cual se espera obtener un pico de LH y FSH, que pueda estimular eficientemente la producción de estradiol. Además, como ya hemos visto, la 17 a-OH-P4 plasmática se eleva en forma excesiva en las pacientes con SOP.
En caso de sospecha de bloqueo enzimático, que no sea evidente por pruebas hormonales basales, se sugiere realizar una prueba de estimulación de la corteza adrenal con ACTH sintética en dosis de 0,25 mg por vía endovenosa, y medir 17a-OH-P4 basal y a la hora. Se ha demostrado que las pacientes con SOP, presentan en su mayoría, mayores niveles de 17a-OH-P4 posterior a las pruebas de estimulación con ACTH sintética y/o agonistas de GnRH, presentándose aproximadamente en el 40-60% de las pacientes con hiperandrogenismo una respuesta excesiva de andrógenos adrenales a la estimulación con ACTH.59
De manera tal que, cuando se evalúe a una paciente con SOP y se solicite al laboratorio su perfil hormonal, y observemos que cualquiera de las hormonas referidas se encuentra sobre sus niveles normales, en ese momento se sospecha la existencia de un SOP de origen adrenal (Tabla II).
RESPUESTA TERAPÉUTICA A LA SUPRESIÓN GLUCOCORTICOIDE EN PACIENTES CON SOP
Las pacientes con SOP consultan, principalmente, por trastornos menstruales, anovulación y/o infertilidad. En cuanto a la terapéutica en este tipo de pacientes, es de suma importancia conocer la participación de la glándula suprarrenal en la patología, debido a que el tratamiento será completamente distinto. Se ha observado que las pacientes anovulatorias, con hirsutismo y elevación de los niveles de andrógenos séricos de origen suprarrenal, no responden bien al tratamiento de inducción de la ovulación con el citrato de clomifeno como monoterapia, y de allí que se fracase en la inducción de la ovulación en estos pacientes con SOP, pues este medicamento no reduce los niveles de andrógenos. En estos casos seleccionados, de pacientes con SOP con contribución adrenal, asociar corticosteroides ayuda a restaurar la ovulación al inhibir la producción suprarrenal de andrógenos. La administración con corticosteroides reduce la estimulación de la ACTH sobre la corteza suprarrenal, lo cual resulta en una disminución de los niveles de cortisol y de ciertos andrógenos.60 Sobre este particular, Daly y col,61 notaron mejoría en la función ovárica de pacientes a los cuales se les inducía la ovulación con citrato de clomifeno, cuando se asociaba dexametasona a las pacientes con niveles básales elevados de DHEAS. Se piensa que esto es debido a la capacidad de inhibir la secreción de ACTH por los glucocorticoides.
Se utiliza dexametasona en dosis de 0,25-1 mg, o sus equivalentes como prednisona (2,5-10 mg) o cortisona (37,5-50 mg). La administración es de preferencia nocturna, con la finalidad de lograr el bloqueo del pico matutino de ACTH y cortisona, y lograr una disminución significativa de los andrógenos en el microambiente folicular, aunque puede repetirse una dosis menor en la mañana, de ser necesario.
Se recomienda evaluar los niveles de testosterona y 17a-OH-P4, como método de seguimiento de este tratamiento, además de la función menstrual.34 Por ejemplo, si se observa que los niveles de testosterona y de DHEA disminuyen sus valores entre los días 21 y 24 del primer ciclo de tratamiento, concomitantemente se miden los valores séricos de P4, para confirmar la ovulación, y a las 8.00 a.m. la concentración plasmática de cortisol, esta última para monitorizar la función suprarrenal. No obstante, esta prueba sólo se realiza en pacientes a las cuales se les administró dexametasona, pues la prednisona exógena reacciona con el ensayo del cortisol. A continuación, en las pacientes con SOP a las cuales se les está induciendo la ovulación, las dosis de citrato de clomifeno pueden incrementarse en los ciclos venideros, si persiste la anovulación, mientras las dosis de glucocorticoides deben reducirse, paulatinamente, de acuerdo si disminuyen los niveles de cortisol, con la finalidad de prevenir los efectos secundarios de los mismos (hipertensión, hiperglicemia, disminución de la respuesta inflamatoria, euforia, hiperlipemia, disminución de la síntesis de proteínas, etc.). No obstante, la inhibición que ejercen los corticosteroides sobre la glándula suprarrenal es temporal, por lo que el riesgo de cambios “cushingoides” es bajo.34 Por otra parte, el uso de glucocorticoides a bajas dosis durante el inicio del embarazo, no se ha relacionado con incremento en la incidencia de complicaciones fetales.34 Es oportuno aclarar, que las pacientes con HAC requieren glucocorticoides de por vida.
RANGO NORMAL DE CONCENTRACIONES HORMONALES SÉRICAS EN LA MUJER ADULTA
| T (ng/mL) | 17 α-OH-P4 (ng/mL) | DEA (ng/mL) | DHEAS (ng/mL) | C (ng/mL) | A4 (ng/mL) |
| <1 | 0,2-0,9 | 2-8 | 400-1200 | 0,05-0,2 | 1-2 |
(T) testosterona. (17 α-OH-P4) 17α-hidroxiprogesterona. (DHEA) dehidroepiandrosterona. (DHEAS) dehidroepiandrosterona sulfato. (C) cortisol. (A4) androstenediona.
Por último, cabe destacar que estas combinaciones terapéuticas de citrato de clomifeno asociados a corticosteroides puede dar tasas de embarazos de alrededor del 60%,62 a diferencia del tratamiento usando solamente al citrato de clomifeno, con el cual aunque el 80% al 90% de las pacientes ovulan, sólo la mitad alcanzan un embarazo.62
REFERENCIAS BIBLIOGRÁFICAS
1. FRANKS S.: Polycystic ovary syndrome: approaching the millennium. Hum Reprod 1997; 12:43-45. [ Links ]
2. AZZIZ R., BLACK V., HINES G.A., FOX L.M., BOOTS L.R.: Adrenal Androgen Excess in the Polycystic Ovary Syndrome: Sensitivity and Responsivity of the Hypothalamic-Pituitary-Adrenal Axis. J Clin Endocrinol Metab 1998; 83:2317-2323. [ Links ]
3. CARMINA E., ROSATO F., JANNI A.: Increased DHEAS levels in PCO syndrome: evidence for the existence of two subgroups of patients. J Endocrinol Invest 1986;9:5-9. [ Links ]
4. MILEWICZ A., SILBER D., MIELECKI P.: The origin of androgen synthesis in polycystic ovary syndrome. Obstet Gynecol 1983;62:601-604. [ Links ]
5. MOLTZ L., SCHWARTZ U., SORENSEN R.: Ovarian and adrenal vein steroids in patients with non-neoplastic hyperandrogenism: selective catetherization findings. Fertil Steril 1984;42:69-75. [ Links ]
6. TERÁN DÁVILA J. La corteza suprarrenal: aspectos embriológicos, morfológicos y funcionales. In: Terán Dávila J, Febres Balestrini F, eds. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca, 1995.p.77-107. [ Links ]
7. BRUHN T., SUTTON R., RIVIER C.L., VALE W.: Corticotropin-releasing factor regulates proopiomelanocortin messenger ribonucleid acid levels in vivo. Neuroendocrinology 1984; 39: 170-175. [ Links ]
8. ANTONI F.A., PALKOVITS M., MAKARA G.B., LINTON E.A., LOWRY P.J., KISS J.: Immunoreactive corticotropin releasing hormone in the hypothalamo-infundibular tract. Neuroendocrinology 1983; 36:815-832. [ Links ]
9. BOLTE E., CAUDERT S., LEFEBRE Y.: Steroid production from plasma cholesterol: II. In vivo conversion of plasma cholesterol to ovarian progesterone and adrenal C-19 and C-21 steroid in humans. J Clin Endocrinol Metab 1974; 38:394-398. [ Links ]
10. HORNSBY P.J., GILL G.N.: Characterization of adult bovine adrenocortical cells throught their life span in tissue culture. Endocrinology 1978; 102:926-936. [ Links ]
11. ORTH D.N., KOVACS W.J., DEBOLD C.R.: Disorders of the adrenal cortex. In: Wilson JD FD, ed. Williams Textbook of Endocrinology. Philadelphia: Editorial Saunders, 1992:489-619. [ Links ]
12. AZZIZ R.: adrenal androgen. In: Adashi EY RJ, Rosenwaks Z,, eds. Reproductive endocrinology, surgery, and technology. Vol. 1161-1180. Philadelphia: Lippincott-Raven Publishers, 1996.p.1162-1180. [ Links ]
13. ROSENFIELD R.: Current concepts of polycystic ovary syndrome. Balliere´s Clin Obstet Gynaecol 1997; 11:303-313. [ Links ]
14. HOFFMAN D.I., KLOVE K., LOBO R.A.: The prevalence and significance of elevated dehydroepiandrosterone sulfate levels in anovulatory women. Fertil Steril 1984; 42:76-81. [ Links ]
15. GÜVEN M., ACBAY O., SULTUYBEK G.: Glucocorticoid receptors on mononuclear leukocytes in polycystic ovary syndrome. Int J Gynaecol Obstet 1998; 63:33-37. [ Links ]
16. INVITTI C., DE MARTIN M., DELITALA G., VELDHUIS J.D., CAVAGNINI F.: Altered morning and nighttime pulsatile corticotropin and cortisol release in polycystic ovary syndrome. Metabolism 1998; 47:143-148. [ Links ]
17. PARKER L.N., ODELL W.D.: Control of adrenal androgen secretion. Endocr Rev 1980; 1:392-410. [ Links ]
18. MCKENNA T., CUNNINGHAM S.: The pathogenesis of adrenal and extraadrenal hyperandrogenism. J Steroid Biochem Mol Biol 1993; 45:117-121. [ Links ]
19. ORENSTEIN H., RASKIND M.A., WYLLIE D., RASKIND W.H., SOULES M.R.: Polysymptomatic complaints and Briquet’s syndrome in polycystic ovary disease. Am J Psychiatr 1986; 143:768-771. [ Links ]
20. GIVENS J.R., WIEDEMANN E., ANDERSEN R.N., KITABCHI A.E.: Beta-endorphin and beta-lipotropin plasma levels in hirsute women: correlation with body weight. J Clin Endocrinol Metab 1980; 50:975-976. [ Links ]
21. LOBO R.A.: The role of the adrenal in polycystic ovary syndrome. Semin Reprod Endocrinol 1984; 2:251-262. [ Links ]
22. D’AMBROGIO G., FACCHINETTI F., GOLINELLI S., SETTI T., PETRAGLIA F., GENAZZANI A.R.: Adrenal steroid responses to naloxone in polycystic ovarian disease. Gynecol Endocrinol 1987; 1:355-361. [ Links ]
23. LOBO R.A., GRANGER L.R., PAUL W.L., GOEBELSMANN U., MISHELL D.R. JR.: Psychological stress and increases in urinary norepinephrine metabolites, platelet serotonin, and adrenal androgens in women with polycystic ovary syndrome. Am J Obstet Gynecol 1983; 145:496-503. [ Links ]
24. KLEITMAN N, HOLZWARTH MA. Catecholaminergic innervation of the rat adrenal cortex. Cell Tissue Res 1985; 241:139-147. [ Links ]
25. TERÁN-DÁVILA J.: Prolactina humana: biosíntesis, regulación neuroendocrina y estados hiperprolactinémicos. En: Terán Dávila J, Febres Balestrini F, eds. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca, 1995. p.487-513. [ Links ]
26. POSNER B.I., KELLY P.A., SHIU R.P.C., FRIESEN H.B.: Studies of insulin, growth hormone and prolactin binding: tissue distribution, species variation and characterization. Endocrinology 1974; 95:521-531. [ Links ]
27. HOMBURG R, ASHKENAZI J, GOLDMAN J. Resistant cases of polycistic ovary disease succesfully treated with a combination of corticosteroids, clomiphene, and bromocriptine. Int Fertil 1988; 33:393-397. [ Links ]
28. LOBO R.A., KLETZKY O.A., KAPTEIN E.M., GOEBELSMANN U.: Prolactin modulation of dehydroepiandrosterone sulfate secretion. Am J Obstet Gynecol 1980; 138:632-636. [ Links ]
29. HANING R.V., JR, CARLSON I.H., FLOOD C.A., HACKETT R.J., LONGCOPE C.: Metabolism of dehydroepiandrosterone sulfate (DS) in normal women and women with high DS concentrations. J Clin Endocrinol Metab 1991; 73:1210-1215. [ Links ]
30. ROSENFIELD R.L.: Mecanismos de hiperandrogenismo en el síndrome de ovarios poliquísticos. Cuadernos de medicina reproductiva 1998;4:59-74. [ Links ]
31. BALEN A.H., DUNGER D.: Pubertal maturation of the internal genitalia (commentary). Ultrasound Obstet Gynaecol 1995; 6:164-165. [ Links ]
32. RITTSMASTER R., DESHWAL N., LEHMAN L.: The role of adrenal hyperandrogenism, insulin resistance and obesity in the pathogenesis of polycystic ovary syndrome. J Clin Endocrinol Metab 1993; 76:1295-1299. [ Links ]
33. DEWAILLY D, CORTET-RUDELLI C, DEROUBAIX-ALLARD D. Markers of abdominal adipose tissue in women: relationship to ovarian function. TEM 1998; 9:68-72. [ Links ]
34. HAMMOND M.G.: Pharmacology of ovulation-inducing drugs. In: Reye WR CR, Rebar RW, Soules MR, eds. Infertility. Evaluation and treatment. Philadelphia: W.B. Saunders Company, 1995: p 127-144. [ Links ]
35. STEINGOLD K., DE ZIEGLER D., CEDARS M.: Clinical and hormonal effects of chronic gonadotropin-releasing hormone agonist treatment in polycystic ovarian disease. J Clin Endocrinol Metab 1987; 65:773-778. [ Links ]
36. TAYLOR A.E.: Understanding the underlying metabolic abnormalities of polycystic ovary syndrome and their implications. Am J Obstet Gynecol 1998; 179:S94-S100. [ Links ]
37. AUCHUS R.J., GELLER D.H., LEE T.C., MILLER W.L.: The regulation of human P450c17 activity: relationship to premature adrenarche, insulin resistance and the polycystic ovary syndrome. TEM 1998; 9:47-50. [ Links ]
38. VICENNATI V., GAMBINERI A., CALZONI F., CASIMIRRI F., MACOR C., VETTOR R., PAS- QUALI R.: Serum leptin in obese women with polycystic ovary syndrome is correlated with body weight and fat distribution but not with androgen and insulin levels. Metabolism 1998; 47:988-992. [ Links ]
39. TEPPA-GARRÁN A.D.: Importancia de la leptina en Ginecología y Obstetricia. Rev Venez Obstet Gynecol 1999;59:35-43. [ Links ]
40. GROSS M.D., WORTSMAN J., SHAPIRO B., MAYERS L.C., WOODBURY M.C.: AYERS J.W.P.: Scintigraphic evidence of adrenal corticoid dysfunction in the polycystic ovary syndrome. J Clin Endocrinol Metab 1986; 62:197-201. [ Links ]
41. EREL C.T., SENTURK L.M., ORAL E., COLGAR U., ERTUNGEALP E.: Adrenal androgenic response to 2-hour ACTH stimulation test in women with PCOS. Gynecol Endocrinol 1998; 12:223-229. [ Links ]
42. GENNARELLI G., HOLTE J., STRIDSBERG M., LUNDQVIST U., MASSOBRIO M., BÄCKSTRÖM T., BERNE C.: Response of the pituitary-adrenal axis to hypoglycemic stress in women with the polycystic ovary syndrome. J Clin Endocrinol Metab 1999; 84:76-81. [ Links ]
43. PARDO-PALMA R.A.: Síndrome de ovarios poliquísticos. En: Terán Dávila J, Febres Balestrini F, editores. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca 1995: 413-459. [ Links ]
44. ABLAN-CANDIA F., TERÁN-DÁVILA J., PARDO-PALMA R.A., FEBRES-BALESTRINI F. En: Terán Dávila J, Febres Balestrini F, editores. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca 1995:351-378. [ Links ]
45. BARNES R.B., ROSENFIELD R.L., BURSTEIN S., EHRMAN D.A.: Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Eng J Med 1989; 320:559-565. [ Links ]
46. GONZALEZ F., CHANG L., HORAB T., STANCZYK F.Z., CRICKARD K., LOBO R.A.: Adrenal dynamic responses to physiologic and pharmacologic adrenocorticotropic hormone stimulation before and after ovarian steroid modulation in women with polycystic ovary syndrome. Fertil Steril 1999; 7:439-444. [ Links ]
47. IBÁÑEZ L., POTAU N., CARRASCOSA.: Insulin resistance, premature adrenarche, and a risk of the polycystic ovary syndrome (PCOS). TEM 1998; 9: 72-76. [ Links ]
48. SIEGEL S.F., FINEGOLD D.N., LANES R., LEE P.A.: ACTH estimulation tests and plasma dehydroepiandrosterone sulfate levels in women with hirsutism. N Eng J Med 1990; 323:849-854. [ Links ]
49. PANG S.: The molecular and clinical spectrum of 3beta-hydroxysteroid dehydrogenase deficiency disorder. TEM 1998; 9:82-87. [ Links ]
50. FEBRES-BALESTRINI F.: Interacción hormonal y fisiológica de la glándula mamaria. En: Terán Dávila J, Febres Balestrini F, eds. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca, 1995.p.109-127. [ Links ]
51. KAHSAR-MILLER M., AZZIZ R.: The development of the polycystic ovary syndrome: family history as a risk factor. TEM 1998; 9:55-58. [ Links ]
52. FRANK S., GHARANI N., WATERWORTH D., BATTY S., WHITE D., WILLIAMSON R., MCCARTHY M.: The genetic basis of polycystic ovary syndrome. Hum Reprod 1997;12: 2641-2648. [ Links ]
53. FRANK S.: Factores genéticos en la etiología del síndrome de ovarios poliquísticos. Cuadernos de medicina reproductiva 1998; 4:45-58. [ Links ]
54. WATERWORTH D.M., BENNETT S.T., GHARANI N.: Linkage and association of insulin gene VNTR regulation polymorphism with polycistic ovary syndrome. Lancet 1997; 349: 986-990. [ Links ]
55. LIU J.H.: Polycystic ovary syndrome variants. Pseudocyesis. In: Adashi EY RJ, Rosenwaks Z, eds. Reproductive Endocrinology, Surgery, and Technology. Philadelphia: Lippincott-Raven Publishers, 1996.p.271-1277. [ Links ]
56. TERÁN-DÁVILA J.: Amenorrea primaria: clasificación, diagnóstico y tratamiento. En: Terán Dávila J, Febres Balestrini F, eds. Endocrinología Ginecológica y Reproducción Humana. Caracas: Ateproca, 1995. p.293-330. [ Links ]
57. BRAY M.A., MUNEYYIRCI-DELALE O., KOFINAS G.D., REYES F.I.: Circadian, ultradian, and episodic gonadotropin and prolactin secretion in human pseudocyesis. Acta Endocrinol 1991;124:501-509. [ Links ]
58. TERÁN-DÁVILA J., FEBRES-BALESTRINI F.: Función hipofisiaria de la pseudociesis. Gac Med Caracas 1991; 99:323-326. [ Links ]
59. SAHIN Y., KELESTIMUR F.: 17-hydroxyprogesterone responses to gonadotrophin-releasing hormone agonist buserelin and adrenocorticotrophin in polycystic ovary syndrome: investigation of adrenal and ovarian cytochrome P450c17a dysregulation. Hum Reprod 1997; 12:910-913. [ Links ]
60. MEIKLE A.W., ODELL W.D.: Effect of short and long-term dexamethasone on 3alfa-androstenediol glucorinide in hirsute women. Fertil Steril 1986; 46:227-231. [ Links ]
61. DALY D.C., WALTERS C.A., SOTO-ALBORS C.E., TOHAN N., RIDDICK D.H.: A randomized study of dexamethasone in ovulation induction with clomiphene citrate. Fertil Steril 1984; 4:844-848. [ Links ]
62. MCCLAMROCK H.D., KATZ E., ADASHI E.Y.: Clomiphene citrate. A 1990 update. Infert Reprod Med NA 1990;1:37-42. [ Links ]
