










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInvestigación Clínica
versión impresa ISSN 0535-5133versión On-line ISSN 2477-9393
Invest. clín v.47 n.4 Maracaibo dic. 2006
Esclerosis Múltiple en niños: clarificando su ubicación dentro del espectro desmielinizante.
Joaquín Antonio Peña1, Cecilia Montiel-Nava1, María Elena Ravelo2, Solmary González1, y Eduardo Mora La-Cruz1
1Postgrado de Neurología Pediátrica, Facultad de Medicina, Universidad del Zulia, Maracaibo, Venezuela y 2Servicio de Neurología, Hospital de Niños J.M. de Los Ríos, Caracas, Venezuela. Correo electrónico: juaco949@hotmail.com
Resumen. La Esclerosis múltiple (EM), es una enfermedad de naturaleza autoinmune debida a la destrucción de la capa de mielina y de las fibras nerviosas y, secundariamente, a un daño neuronal progresivo. Se caracteriza por la ocurrencia sucesiva de focos de desmielinización diseminados en tiempo y espacio en varias áreas de la sustancia blanca del SNC, incluyendo la región peri ventricular, médula espinal, tronco encefálico, cerebelo y nervio óptico. Como el diagnóstico en edad pediátrica es mucho más difícil debido a la mayor frecuencia de pacientes con Encefalomielitis Aguda Diseminada (EMAD), y al mayor reto que enfrenta el médico para tratar de excluir otras entidades clínicas, se puede establecer este diagnóstico cuando se demuestra la ocurrencia de lesiones en la sustancia blanca con diseminación en tiempo y espacio las cuales no pueden ser explicadas por otros mecanismos o entidades patológicas. El propósito de este artículo es revisar los parámetros actuales de la EM en edad pediátrica, los dilemas en cuanto a la validez de los criterios para el diagnóstico, manifestaciones clínicas, su diferenciación de otros trastornos desmielinizantes, y su proceso diagnóstico. La EM, aunque infrecuente, es un diagnóstico válido dentro del espectro de las enfermedades desmielinizantes inflamatorias de la infancia. El cuadro clínico puede ser indistinguible de una encefalopatía aguda diseminada multifocal o puede presentarse con manifestaciones focales. Para confirmar o descartar el diagnóstico se requiere un razonado juicio clínico y la práctica de exámenes paraclínicos. No es posible la diferenciación entre la EMAD y la EM en un primer episodio ni por la clínica, el líquido céfalo raquídeo (LCR) o la neuroimagen; siendo aún necesarios criterios de consenso tanto clínicos como paraclínicos. Están pendientes todavía muchas interrogantes las cuales sólo pueden responderse mediante los estudios prospectivos, usando medidas clínicas uniformes que permitan delinear los rasgos demográficos, neurológicos, y neuropsicológicos de la EM y las otras formas de trastornos desmielinizantes adquiridos de la infancia
Palabras clave: Esclerosis Múltiple, Encefalomielitis Aguda Diseminada, enfermedades desmielinizantes, neuroimagen
Multiple Sclerosis in children: clarifying its place among the demyelinating spectrum.
Abstract. Multiple sclerosis (MS) is an autoimmune disease caused by the destruction of the myelin layer and the nervous fibers, and secondary by a progressive neuronal damage. It is characterized by episodes of demyelination disseminated in time and space in different areas of the white matter of the CNS which includes periventricular region, spinal cord, brain stem, cerebellum and optical nerve. Due to the confusing differential diagnosis of MS in children with other demyelinating diseases such as ADEM, it is important to reach this diagnosis when there is proof of white matter lesions disseminated in time and space that cannot be explained by any other mechanisms or pathologies. The goal of this paper is to review the diagnostic parameters used for MS in the pediatric age, the dilemmas regarding the validity of diagnostic criteria, clinical manifestations, differentiation of other demyelinating diseases, and the diagnostic process. MS although infrequent, is a valid diagnosis among the spectrum of childhood inflammatory demyelinating diseases. The clinical presentation might be indistinguishable from a multifocal acute disseminated encephalopathy or could be presented with just focal signs. A reasonable clinical judgment and the practice of laboratory tests confirm or rule out the diagnosis. It is not possible to differentiate between ADEM and MS in a first episode, nor by the clinical, the CSF, neither the neuroimaging. There are still needed consensus criteria both clinical and laboratory test. There are many question still to be answered using prospective studies, and standardized clinical measures that will allow the delimitation of the demographic, neurological, and neuropsychological aspects of the MS and other form of acquired demyelinating diseases in children.
Key words: Multiple Sclerosis, Acute disseminated encephalomyelitis, demyelinating syndromes, neuroimaging.
Recibido: 04-02-2006 Aceptado: 11-05-2006
INTRODUCCIÓN
La Esclerosis múltiple (EM), es una enfermedad de naturaleza autoinmune cuyo origen se relaciona con la destrucción de la capa de mielina y de las fibras nerviosas y, secundariamente, a un daño neuronal progresivo. Se caracteriza por la ocurrencia sucesiva de focos de desmielinización diseminados en tiempo y espacio en varias áreas de la sustancia blanca del SNC, incluyendo la región periventricular, médula espinal, tronco encefálico, cerebelo y nervio óptico (1). La EM representa una de las entidades más comunes entre los individuos jóvenes, y su diagnóstico es infrecuente en la edad pediátrica. Se han obtenido grandes avances respecto del diagnóstico y manejo de la EM en el adulto (2-4). La mayor disponibilidad de las investigaciones paraclínicas: potenciales evocados, inmunoglobulinas y bandas oligoclonales en el LCR y, en particular, la resonancia magnética cerebral, han facilitado su diagnóstico (5). Así mismo, se ha enfatizado recientemente el valor de un diagnóstico temprano y tratamiento adecuado, habida cuenta de los avances en las terapias efectivas que alteran el curso de la enfermedad. Como el diagnóstico en edad pediátrica es mucho más difícil debido a la mayor frecuencia de pacientes con Encefalomielitis aguda diseminada (EMAD), y al mayor reto que enfrenta el médico para tratar de excluir otras entidades clínicas como posibles diagnósticos, se puede establecer este diagnóstico sólo cuando se demuestra la ocurrencia de lesiones en la sustancia blanca con diseminación en tiempo y espacio, las cuales no pueden ser explicadas por otros mecanismos o entidades patológicas (6). Si bien actualmente existen discrepancias en torno a la definición de EMAD, asimismo se presentan dudas sobre la validez de los criterios necesarios para los pacientes pediátricos con sospecha de EM, o si un síndrome clínicamente aislado puede diferenciarse de la EMAD y, no menos importante, si la EMAD recidivante puede distinguirse de la EM (7). Tampoco se han identificado completamente los factores de riesgo ni el papel de los factores étnicos o genéticos en la población pediátrica. No obstante, la observación de niños con un cuadro clínico sugestivo, asociado con pruebas de laboratorio o neuroimagen como las mencionadas en la población adulta con EM, hace necesario aplicar esquemas diagnósticos que combinen los hallazgos clínicos y paraclínicos, y permitan establecer el diagnóstico en este grupo de pacientes. El propósito de este artículo es revisar los parámetros actuales de la EM en edad pediátrica, los dilemas en cuanto a la validez de los criterios para el diagnóstico, manifestaciones clínicas, su diferenciación de otros trastornos desmielinizantes, y su proceso diagnóstico.
EPIDEMIOLOGÍA
La prevalencia de la EM es variable, pero se considera actualmente que el riesgo de la población general es de 1 en 1000. Se estima una población mundial de 2,5 millones de personas; 450.000 han sido diagnosticados en los Estados Unidos y 50.000 en Canadá (8). La EM afecta preferentemente a los sujetos entre los 20 y 40 años, con un pico máximo a los 30 años, predominando en las mujeres con una proporción de 1,8 a 2. Los estudios de población y series de casos control muestran que entre 2,5 a 5% de la población con EM son menores de 17 años (4, 9-14). La edad media de presentación para estos pacientes es de 13 años. En Venezuela, del total de 714 pacientes registrados en el programa nacional de EM, 15 son menores de 17 años, lo cual representa un 2,5% (15). Se han publicado casos anecdóticos en sujetos menores de un año y una serie de 49 pacientes menores de 6 años (16). Se estima que aproximadamente el 0,3 a 2% del total de pacientes con EM es menor de 10 años, si bien esta proporción no incluye los sujetos con presentación clínica atípica, que pudieran no ser diagnosticados (13, 17, 18). En los sujetos mayores de 12 años predominan las mujeres en una relación de 3:1, a diferencia de los menores de 10 años con una relación de 1:1, enfatizando el papel de posibles factores hormonales.
INTERROGANTES AÚN PENDIENTES
Al no existir un marcador biológico específico, algunos de los grandes dilemas que se le presentan al clínico son los referentes a: 1) ¿Cuáles criterios son necesarios para el diagnóstico en pacientes pediátricos con sospecha de EM? 2) ¿Cuál debe ser la aproximación clínica ante un paciente que presenta un primer episodio de disfunción neurológica focal que pudiera corresponder a la primera manifestación de EM? y 3) ¿Cómo diferenciar una forma de enfermedad desmielinizante inflamatoria aguda, como la EMAD, de la EM? (19, 20).
Si bien el diagnóstico de EM en niños es posible sobre la base de elementos clínicos, paraclínicos y evolutivos que demuestran la diseminación temporal y espacial del trastorno, debe señalarse que en ocasiones se confrontan problemas para el diagnóstico correcto y sólo la evolución en el tiempo da la respuesta a esta incógnita. Cabe mencionar también los aspectos madurativos del SNC, relacionados con la expresión clínica del trastorno en la edad pediátrica. Este hecho añade una particular consideración con relación a los criterios de neuroimagen, los cuales no han sido validados en la población pediátrica, razón por la cual no hay consenso (20-21).
A pesar de que cada día se acumula mayor evidencia acerca de la validez de la EM en niños, existen dificultades al momento de diferenciar esta entidad clínica de las otras enfermedades con clínica y neuroradiología parecida. A continuación, se analizan cada una de estas interrogantes relacionadas con la EM y su presentación en edades pediátricas.
1) Criterios necesarios para edades pediátricas. Aproximación al diagnóstico
El diagnóstico de EM se basa en la demostración de los elementos clínicos que son la evidencia de una disfunción de la sustancia blanca, la cual debe comprometer más de un área del SNC y debe ser diseminada en tiempo. Deben excluirse otras explicaciones posibles de los síntomas y descartar otras entidades, para lo cual es necesario obtener información adicional suministrada por una adecuada historia clínica y examen neurológico detallado. Este último comprende la búsqueda de los signos que sugieren alteraciones del nervio óptico, la reducción de la fuerza, alteración de los reflejos, coordinación, y sensibilidad. Un seguimiento clínico y la práctica de otros exámenes permiten orientar el diagnóstico. Como no existen marcadores paraclínicos patognomónicos de la EM, el diagnóstico es principalmente clínico con la ayuda de esquemas diagnósticos que combinan los hallazgos clínicos y paraclínicos (2, 22-24).
La presentación clínica de la EM en niños es parecida a la encontrada en adultos, con algunas especificidades. Las manifestaciones iniciales pueden ser monosintomáticas o poli sintomáticas, e incluyen signos y síntomas sensitivos o visuales. También pueden observarse síntomas motores y en general, aquellos que sugieren compromiso de tallo cerebral. Si bien los signos y síntomas de presentación en niños son similares a los del adulto, predominan los trastornos visuales, la afectación sensitiva, la ataxia, y trastornos de conciencia en grados variables (6, 9, 24). La paresia motora pura que compromete típicamente los miembros, es un síndrome de presentación menos común en adolescentes (15%) que en adultos (50%). En dos series importantes, los síntomas clínicos iniciales consistieron principalmente en manifestaciones sensitivas, motoras y de tallo cerebral (9,10). Por otra parte, en niños prepuberales es frecuente que se manifieste en forma aguda como una encefalopatía difusa, o una encefalopatía de tronco con deterioro del sensorio, meningismo y compromiso de pares craneanos, hecho que obliga, de entrada, a considerar como diagnóstico diferencial inicial con la EMAD (6, 25).
En una serie de 57 pacientes pediátricos, 46 con diagnóstico de EMAD y 11 de EM, se encontró que la edad de los sujetos con EM era mayor (promedio = 10,73 años) que los que presentan EMAD (promedio = 5,78 años), siendo esta diferencia significativa (p = 0,001). Hubo mayor número de varones en ambas entidades clínicas; en la EMAD la proporción fue de 1:2, y en la esclerosis múltiple de 1:1, favorable a los varones. Al estudiar los síntomas de presentación de estos pacientes (Tabla I). Se observó una diferencia significativa en los síntomas iniciales de ambas entidades clínicas. En específico, se puede ver como la fiebre y los antecedentes infecciosos son significativos para la EMAD, más no para la EM. Por el contrario, las alteraciones visuales son significativas en los niños con EM. De los casos con diagnóstico inicial de EMAD que evolucionaron hacia una EM, todos tuvieron como síntoma de presentación inicial alteraciones visuales (neuritis óptica y/o diplopía). Los otros síntomas que incluyen convulsiones, ataxia, paresias, y parálisis facial, no fueron discriminativos entre ambas muestras (26).
SÍNTOMAS INICIALES EN PACIENTES CON ENFERMEDADES DESMIELINIZANTES (EMAD Y EM)
| EMAD n | n = 46 % | EM | n = 11 % | X2 | P | |
| Antecedentes infecciosos | 26 | 56,52 | 0 | 0 | 11,43 | 0,001 |
| Fiebre | 28 | 60,87 | 8,33 | 8,33 | 9,523 | 0,002 |
| Convulsiones | 25 | 54,35 | 25 | 25 | 2,6 | 0,1 |
| Ataxia | 5 | 10,89 | 8,33 | 8,33 | 0,038 | 0,846 |
| Paresias | 22 | 47,83 | 33,33 | 33,33 | 4,42 | 0,11 |
| Alteraciones visuales | 5 | 10,89 | 58,33 | 58,33 | 14,871 | 0,001 |
| Parálisis facial | 5 | 10,89 | 0 | 0 | 1,311 | 0,252 |
Así se tiene un punto de orientación diagnostica: en un niño con elementos clínicos que sugieren disfunción de la sustancia blanca, y con antecedentes infecciosos y fiebre se podría pensar desde un inicio en una EMAD. Si por el contrario, estos signos no están presentes pero si hay alteraciones visuales, esto sería un punto para inclinar el diagnóstico hacia una EM, o al menos realizar un seguimiento riguroso teniendo presente la posibilidad de una recaída.
La forma recurrente-remitente de la EM es la evolución más común en la edad pediátrica, especialmente en los menores de 10 años, aunque no suele mantener una evolución progresiva luego de la pubertad. Se ha señalado que los casos con inicio posterior (después de los 10 años), muestran un curso progresivo continuado, pero se necesitan estudios prospectivos para poder confirmar esta observación (6, 7). En la serie de Ghezzi y col. (10) 34% de los pacientes presentaron recaídas en el primer año; 6 de los 7 (87%) pacientes descritos por Sánchez Calderón y col. (13) evolucionaron en forma recurrente-remitente y el 63% de los niños menores de 6 años descritos por Ruggieri y col. (16), tuvieron recaídas.
Si existe la sospecha clínica de EM, la Resonancia magnética (RM) se emplea como auxiliar en la certeza diagnóstica o para predecir el desarrollo de EM en los pacientes con síndromes clínicamente aislados (primer episodio de desmielinización). No obstante, se desconoce el valor pronóstico de la RM inicial para recaídas en edad pediátrica y su papel para diferenciar con seguridad entre una enfermedad monofásica y recurrente (19). Los hallazgos de neuroimagen consisten en focos de desmielinización particularmente visibles en las secuencias ponderadas en T2 y FLAIR, principalmente bajo la forma de hiperseñal. Se trata de lesiones de diámetro variable, hiperintensas en secuencias T2, de distribución multifocal, con coexistencia de placas activas o inflamatorias y placas crónicas o inactivas, lo cual confirma la diseminación en tiempo del trastorno (27-28). La tendencia centrofórica de las placas, localizadas preferentemente en la sustancia blanca profunda, cuerpo calloso, tallo cerebral, y la zona periventricular, es un carácter definido de la EM que suele distinguirla de la EMAD. La diferencia temporal se establece completando el estudio con gadolinio, el cual permite visualizar el grado de reforzamiento de las lesiones (18, 24). En estudios de seguimiento, se pueden encontrar nuevas lesiones en pacientes con EM. Es importante enfatizar que estos estudios han permitido comprender que la aparición de ciertos síntomas neurológicos focales en pacientes con riesgo epidemiológico elevado, obliga a investigar la posibilidad de un primer episodio de la enfermedad. En dichos casos, es altamente recomendable practicar una serie de pruebas, incluyendo la resonancia magnética cerebral y/o de médula espinal (5).
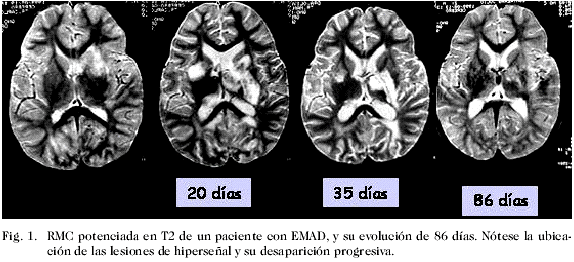
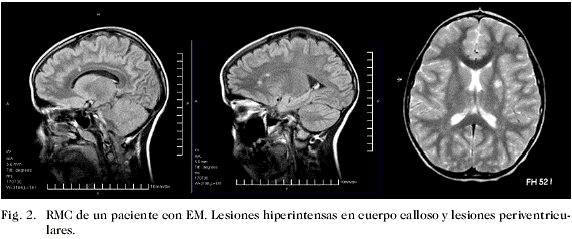
Los criterios diagnósticos propuestos por McDonald y col. (23), recientemente revisados por Polman y col. (29), incorpora la RM para documentar la diseminación en espacio (áreas múltiples de desmielinización) y tiempo (aparición de nuevas lesiones en los estudios de seguimiento). Otros criterios utilizados en neuroimagen de la EM son los de Paty y col. (22) y los de Fazekas y col. (30). Como se señaló, ninguno de los criterios imagenológicos, incluyendo los de McDonald y col., han sido validados en la población pediátrica; por tanto, se hace necesario un consenso sobre las características radiológicas de la EM en este grupo de pacientes (21, 29-32). En un estudio piloto en el cual se investigaron los criterios de Paty y col (22), Fazekas y col. (30), McDonald y col. (23) en una cohorte de 20 niños con un primero y segundo evento desmielinizante, pudo evaluarse la aplicabilidad de estos parámetros. La mayor proporción de pacientes cumplía los criterios de Paty y col. y Fazekas y col. (22, 30). Sólo un 53% de los niños cumplió los criterios de McDonald y col. (23) al momento del primer episodio, y tuvieron también menos lesiones en la sustancia blanca con respecto a los adultos con diagnóstico reciente de EM. Los autores sugieren que los niños con EM están más cerca del momento biológico verdadero de la enfermedad y, como consecuencia, han tenido menos tiempo para acumular lesiones de la sustancia blanca. Señalan asimismo, que la EM en niños comienza antes de finalizar la mielinización, hecho que incide en la expresión estructural de las lesiones (21). Por esta razón, los autores recomiendan estudios prospectivos y controlados que permitan desarrollar criterios mejor ajustados a la población pediátrica. Debe enfatizarse de nuevo el valor de la neuroimagen para la identificación de los niños con enfermedades inflamatorias desmielinizantes, incluyendo la EM. Aunque esta puede ser normal, especialmente en las fases iniciales del trastorno, es sin duda el método más sensible para el diagnóstico, y las anomalías objetivadas son esencialmente iguales a las descritas en adultos. La Fig. 1 muestra la evolución documentada por RM de un paciente con EMAD, siendo evidente la ubicación característica de las lesiones hiperintensas con una progresiva disminución de la señal de intensidad y del tamaño de las mismas. Por el contrario, en la Fig. 2 correspondiente a un sujeto con EM, se observan lesiones menos extensas, mejor delimitadas, frecuentemente ubicadas en la sustancia blanca periventricular, que suelen permanecer más tiempo y se asocian con nuevas lesiones en los estudios de control (diseminación en espacio).
En ocasiones, se necesita una evidencia adicional que demuestre la ocurrencia de más de un ataque. Por ejemplo, en un niño que ha experimentado un sólo episodio de desmielinización o sólo ha presentado un síntoma, la respuesta anormal de ciertas pruebas ofrece la evidencia de una segunda área de desmielinización en el cerebro. Si bien algunos hallazgos en el LCR (hiperproteinorraquia o discreta pleocitosis linfocítica) no son exclusivos para la EM, pueden detectarse alteraciones muy sugestivas como las bandas oligoclonales o demostrarse evidencia de síntesis intratecal de IgG (5, 6, 19). También, los potenciales evocados han sido incorporados en los criterios diagnósticos con el fin de ofrecer el soporte para el diagnóstico de la EM. Estos pueden objetivar lesiones subclínicas y permiten establecer la naturaleza diseminada del trastorno.
Es razonable entonces concluir que, al igual que en el adulto, es posible el diagnóstico de EM en edades pediátricas siguiendo los esquemas que combinan los hallazgos clínicos y paraclínicos. Así, son suficientes argumentos en favor de la EM: a) la presencia de dos o más ataques clínicos que comprometen dos áreas separadas del SNC, b) la confirmación de signos y síntomas en el momento, sea por que se demuestre un ataque previo por los hallazgos del examen o por una historia de síntomas previos con evidencia confirmatoria en las pruebas paraclínicas y c) la constatación de signos de desmielinización en la RM.
2. ¿Cuál debe ser la aproximación clínica ante un paciente pediátrico que presenta un primer episodio de disfunción neurológica focal que pudiera corresponder a la primera manifestación de EM?
Algunos síntomas específicos como la neuritis óptica han sido analizados en un estudio multicéntrico con seguimiento de 5 años. Los resultados, basados en pacientes principalmente adultos, sugieren que aquellos sujetos con alteraciones en la RM presentan un riesgo de hasta 50% de desarrollar la enfermedad en un lapso de 5 años (33). Otros estudios, en los cuales se han usado los interferones, revelaron que los pacientes con síntomas visuales (neuritis óptica), del tallo cerebral, o médula espinal tenían riesgo elevado de desarrollar la enfermedad en un período de 1-2 años si presentaban más de 2-3 lesiones en la RM localizadas en áreas cerebrales diferentes a las que determinaban sus síntomas iniciales (34, 35). Estos trabajos ofrecen evidencia importante que obligan a considerar la necesidad de estudio y seguimiento de los pacientes con un primer episodio desmielinizante, por la posibilidad de que se esté en presencia de un primer episodio de EM. Se impone entonces la práctica de exámenes paraclínicos para confirmar o descartar el diagnóstico. Para apoyar el proceso diagnóstico, nuestro grupo de trabajo ha desarrollado un árbol de decisión que toma en consideración los diferentes elementos para alcanzar un buen diagnóstico (Fig. 3). Ante un paciente con enfermedad neurológica de etiología desconocida, con síntomas principalmente de afectación visual, medular o de sustancia blanca, focal o multifocal, sin antecedentes de enfermedad neurológica previa, se plantea el diagnóstico de enfermedad inflamatoria desmielinizante. La normalidad o no de la RM, permitirá el planteamiento del tipo de trastorno que será corroborado con las otras pruebas paraclínicas (LCR, potenciales evocados).

3. ¿Cómo diferenciar una forma de enfermedad desmielinizante inflamatoria aguda, como la EMAD, de la EM?
Con relación a la EM, la EMAD suele ser más frecuente en niños, y los signos y síntomas de encefalopatía aguda pueden aparecer de forma espontánea sin aparentes signos de infección previa (36). Hay diferencias entre ambas respecto de la presentación clínica, pero deben recordarse las presentaciones “encefalíticas” de la EM, frecuentemente observadas en niños. Se señala también que entre el 15 y 35% de los pacientes con diagnóstico inicial de EMAD, evoluciona a una enfermedad multifásica con recaídas y compatible con EM (6, 36, 37). Cuando la EMAD recurre no es posible su diferenciación de la EM sobre la base de los hallazgos clínicos, del LCR o de la neuroimagen. En este respecto, hay una terminología confusa de estas formas multifásicas y recurrentes y se atribuye al mismo trastorno una nomenclatura diferente; bifásica, multifásica, recurrente, recidivante, en recaída, entre otros. Es ampliamente reconocido que algunos niños con EMAD desarrollan episodios remotos de desmielinización con localización diferente a la del episodio inicial; otros en cambio, presentan un solo episodio y, finalmente, otros sujetos presentan episodios recurrentes con localización similar a la presentación inicial. Por lo general las recurrencias de la EMAD con distribución anatómica, signos y síntomas similares y en proximidad temporal habitualmente dentro de 3 meses del episodio inicial, después de la resolución parcial o completa de aquellas anomalías clínicas o de neuroimagen representan la encefalomielitis diseminada multifásica (EMDM), que ocurren dentro del mismo proceso agudo monofásico (37). Otros autores consideran la EM cuando las recaídas acaecen pasados 6 meses (37-39). En este sentido, Dale y col. (38), señalan que la recaída de la EMAD debe presentarse en un lapso no mayor de 8 semanas tras la suspensión de los esteroides. No obstante, otros autores consideran la EMDM, casos bifásicos, con posibles recurrencias con lapsos de 2 y 8 años después del episodio inicial (18, 40).
Un criterio principal para la orientación diagnóstica de la EMAD parece ser la falta de diseminación en el tiempo (6). La diferenciación entre los signos tempranos de EM y EMAD sólo es posible cuando se hace un seguimiento clínico prolongado, que permita la observación de recaídas. Casi ninguna de las definiciones de EMAD considera la diseminación en el tiempo como un criterio excluyente, si bien las recurrencias son señaladas con frecuencia. Esta observación ha motivado que algunos autores consideren el diagnóstico de EM si la recaída no ocurre dentro de las primeras semanas del episodio inicial. Es por ello importante estudiar la diferenciación de la EMAD y la EM e identificar, a partir del primer evento desmielinizante, los factores de riesgo y sus influencias para la aparición de recaídas. Algunos estudios han comparado la EMAD y la EM definiendo algunos elementos clínicos y de neuroimagen que son propios de uno u otro trastorno. Así, la existencia de signos generales, como alteración de la conciencia, convulsiones, cefalea, fiebre y meningismo son observados más frecuentemente en la EMAD y con menor frecuencia en la EM (17, 41-42) Los hallazgos de la RM ya comentados, con numerosas lesiones, no bien delimitadas y predominio en los ganglios basales son más frecuentemente objetivados en la EMAD. En un estudio reciente con una cohorte de 296 niños con un primer evento desmielinizante, se intentó analizar los factores pronóstico para una segunda recaída. El diagnóstico inicial comprendía EMAD, EM, o episodio focal (neuritis óptica, mielitis transversa o disfunción de tallo cerebral). Los autores consideraron que si el segundo ataque no ocurría dentro del primer mes de la enfermedad se catalogaría como EM. Al final del estudio, que tuvo un seguimiento promedio de 3 años, 57% de los niños cumplió criterios para el diagnóstico final de EM y 29% para EMAD (un solo episodio). Del total de pacientes con EM 20%, fueron considerados tener un diagnóstico inicial de EMAD (7).
En una serie con niños menores de 6 años diagnosticados de EM, 10 de 28 sujetos tenían un diagnostico inicial de EMAD (6). Así, en el momento actual no es posible la diferenciación con certeza entre la EMAD y la EM, y se necesitan mayores estudios epidemiológicos que analicen los factores de riesgo para la ocurrencia de recaídas después de un primer evento. Parece razonable incluir en un mismo espectro de enfermedades desmielinizantes inflamatorias del SNC, los eventos aislados como la neuritis óptica, la mielitis transversa, las variantes monofásicas y multifásicas de la EMAD, siendo cada una de ellas potencialmente capaces de representar las manifestaciones iniciales de la EM en la infancia. Una contribución importante para aclarar esta interrogante sería la obtención de un marcador biológico que pueda predecir a partir de un primer episodio de disfunción neurológica como la EMAD, cuáles pacientes tienen alto riesgo de cursar con evoluciones multifásicas.
CONCLUSIONES
La EM, aunque infrecuente, es un diagnóstico válido dentro del espectro de las enfermedades desmielinizantes inflamatorias de la infancia. El cuadro clínico puede ser indistinguible de una encefalopatía aguda diseminada multifocal o puede presentarse con manifestaciones focales. Un razonado juicio clínico y la práctica de exámenes paraclínicos confirman o descartan el diagnóstico. No es posible la diferenciación entre la EMAD y la EM en un primer episodio ni por la clínica, el LCR o la neuroimagen. Se necesitan criterios de consenso tanto clínicos como paraclínicos. Están pendientes todavía muchas interrogantes, las cuales sólo pueden responderse mediante los estudios prospectivos, usando medidas clínicas uniformes que permitan delinear los rasgos demográficos, neurológicos, y neuropsicológicos de la EM y las otras formas de trastornos desmielinizantes adquiridos de la infancia. Finalmente, se debe pensar en el diagnóstico de EM cuando los signos clínicos al inicio involucran más de una modalidad neurológica, con afectación visual, sensitiva, motora, de tallo cerebral, e incluso con manifestaciones atípicas de etiología poco clara. No se debe, por tanto, negar su existencia sino estudiarla en profundidad, y así continuar aclarando estas interrogantes o tal vez enfrentarse a otras novedosas.
REFERENCIAS
1. Sluder JA, Newhouse P, Fain D. Pediatric and adolescent multiple sclerosis. Adolesc Med 2002; 13(3):461-485. [ Links ]
2. Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, Johnson KP, Sibley WA, Silberberg DH, Tourtellotte WW. New diagnostic criteria for multiple sclerosis: Guideline for research protocols. Ann Neurol 1983; 13:227-231. [ Links ]
3. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983; 33:1444-1452. [ Links ]
4. Boiko A, Vorobeychik G, Paty D, Devonshire V, Sadovnick D. University of British Columbia MS Clinic Neurologists. Early onset multiple sclerosis: a longitudinal study. Neurology 2002; 59:1006-1010. [ Links ]
5. Cohen J, Rensel M. The Differential Diagnosis and clues to Misdiagnosis. En: Burks J, Johnson K (eds.) Multiple Sclerosis. New York. Demos 2000:127-38. [ Links ]
6. Mikaeloff Y, Tardieu M. Sclérose en plaques chez l énfant. Journal de Pédiatrie et de Puériculture 2003; 16:401-405. [ Links ]
7. Mikaeloff Y, Suissa S, Vallee L, Lubetzki C, Ponsot G, Confavreux C, Tardieu M; KIDMUS Study Group. First Episode of Acute CNS Inflammatory Demyelination in Childhood: Prognostic Factors for Multiple Sclerosis and Disability. J Pediatr 2004; 144:246-252. [ Links ]
8. Ebers GC, Sadovnick AD. Epidemiology. En: Paty DW, Ebers GC (Eds.) Multiple Sclerosis, Philadelphia. FA Davis, 1998:5-28. [ Links ]
9. Duquette P, Murray TJ, Pleines J, Ebers GC, Sadovnick D, Weldon P, Warren S, Paty DW, Upton A, Hader W. Multiple sclerosis in childhood: clinical profile in 125 patients. J Pediatr 1987; 111(3):359-363. [ Links ]
10. Ghezzi A, Deplano V, Faroni J, Grasso MG, Liguori M, Marrosu G, Pozzilli C, Simone IL, Zaffaroni M. Multiple sclerosis in Childhood. Clinical features of 149 cases. Mult scler 1997; 3:43-46. [ Links ]
11. Roulet E. La sclerose en plaques chez l’enfant. Rev Neurol 1998; 154: 619-622. [ Links ]
12. Boutin B, Esquivel E, Mayer M, Chaumet S, Ponsot G, Arthuis M. Multiple sclerosis in children: report of clinical and paraclinical features of 19 cases. Neuropediatrics 1988; 19:118-23. [ Links ]
13. Sánchez-Calderon M, de Santos T, Martin S, Angulo T, Careaga J, Campos-Castello J. Esclerosis múltiple en la infancia: nuestra experiencia y revisión de la literatura. Rev Neurol 1998; 27:237-241. [ Links ]
14. Simone IL, Carrara D, Tortorella C, Liguori M, Lepore V, Pellegrini F, Bellacosa A, Ceccarelli A, Pavone I, Livrea P. Course and prognosis in early- onset multiple sclerosis: comparison with adult-onset forms. Neurology 2002; 59:1922-1928. [ Links ]
15. Peña J. Esclerosis múltiple en niños. Invest Clin 2004; 45(Supp1):24-27. [ Links ]
16. Ruggieri M, Polizzi A, Pavone L, Grimaldi LM. Multiple sclerosis in children under six years of age. Neurology 1999; 53:478-484. [ Links ]
17. Dale RC, de Sousa C, Chong WK, Cox TC, Harding B, Neville BG. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 2000; 123: 2407-2422. [ Links ]
18. Hynson JL, Kornberg AJ, Coleman LT, Shield L, Harvey AS, Kean MJ. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology 2001; 56:1308-1312. [ Links ]
19. Gadoth N. Multiple sclerosis in children. Brain & Dev 2003; 25:229-232. [ Links ]
20. Mikaeloff Y, Adamsbaum C, Husson B, Vallee L, Ponsot G, Confavreux C, Tardieu M, Suissa S; KIDMUS Study Group on Radiology. MRI prognostic factors for relapse after acute CNS inflammatory demyelination in childhood. Brain 2004; 127:1942-1947. [ Links ]
21. Hahn CD, Shroff MM, Blaser SI, Banwell BL. MRI criteria for multiple sclerosis: Evaluation in a pediatric cohort. Neurology 2004; 62:806-808. [ Links ]
22. Paty DW, Oger JJ, Kastrukoff LF, Hashimoto SA, Hooge JP, Eisen AA, Eisen KA, Purves SJ, Low MD, Brandejs V. MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 1988; 38:180-185. [ Links ]
23. McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, McFarland HF, Paty DW, Polman CH, Reingold SC, Sandberg-Wollheim M, Sibley W, Thompson A, van den Noort S, Weinshenker BY, Wolinsky JS. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001; 50: 121-127. [ Links ]
24. Tenembaum S, Fejerman N. Enfermedades desmielinizantes. En: Fejerman N, Fernández-Alvarez E, (eds.) Neurología pediátrica, 2nd ed. Buenos Aires: Panamericana 1997: 929-940. [ Links ]
25. López-Pison J, Garcia-Bodega O, Díaz-Suárez M, Bajo-Delgado AF, Cabrerizo-de Diago R, Pena-Segura JL. Inflamación diseminada episódica del sistema nervioso central en niños. Revisión casuística de un período de 13 años. Rev Neurol 2004; 38:405-410. [ Links ]
26. Peña JA. Montiel-Nava C, Ravelo ME, González S, Mora La Cruz E. Esclerosis múltiple en niños. Problemas específicos de diagnóstico. (in extenso) XIII Congreso de la Asociación Iberoamericana de Neurología en Philadelphia, Pennsylvania, 2005, March 31. [ Links ]
27. Brass SD, Caramanos Z, Santos C, Dilenge ME, Lapierre Y, Rosenblatt B. Multiple sclerosis vs. acute disseminated encephalomyelitis in childhood. Pediatr Neurol 2003, 29:227-231. [ Links ]
28. Kesselring J, Miller DH, Robb SA, Kendall BE, Moseley IF, Kingsley D, du Boulay EP, McDonald WI. Acute disseminated encephalomyelitis and MRI findings and the distinction from multiple sclerosis. Brain 1990, 113:291-302. [ Links ]
29. Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, Lublin FD, Metz LM, McFarland HF, O’Connor PW, Sandberg-Wollheim M, Thompson AJ, Weinshenker BG, Wolinsky JS. Diagnostic Criteria for Multiple Sclerosis: 2005 Revisions to the “McDonald Criteria”. Ann Neurol 2005; 58:840-846. [ Links ]
30. Fazekas F, Barkhof F, Filippi M, Grossman RI, Li DK, McDonald WI, McFarland HF, Paty DW, Simon JH, Wolinsky JS, Miller DH. The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis. Neurology 1999; 53(3):448-456. [ Links ]
31. Tintore M, Rovira A, Martinez MJ, Rio J, Diaz-Villoslada P, Brieva L, Borras C, Grive E, Capellades J, Montalban X. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. Am J Neuroradiol 2000; 21(4):702-706. [ Links ]
32. Tintore M, Rovira A, Rio J, Nos C, Grive E, Sastre-Garriga J, Pericot I, Sanchez E, Comabella M, Montalban X. New diagnostic criteria for multiple sclerosis: application in first demyelinating episode. Neurology. 2003; 60:27-30. [ Links ]
33. Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, Comi G, Ader HJ, Losseff N, Valk J. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain. 1997; 120 (Pt 11): 2059-2069. [ Links ]
34. Beck RW, Cleary PA, Trobe JD, Kaufman DI, Kupersmith MJ, Paty DW, Brown CH. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med 1993; 329:1764-69. [ Links ]
35. Jacobs LD, Beck RW, Simon JH, Kinkel RP, Brownscheidle CM, Murray TJ, Simonian NA, Slasor PJ, Sandrock AW. Intramuscular Interferon Beta 1 a therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med 2000; 343:898-904. [ Links ]
36. Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernandez O, Hartung H, Seeldrayers P, Sorensen PS, Rovaris M, Martinelli V, Hommes OR. Early Treatment of Multiple Sclerosis Study Group. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomized study. Lancet 2001; 357:1576-82. [ Links ]
37. Peña JA, Montiel-Nava C, Hernandez F, Medrano E, Valbuena O, Cardozo J. Encefalomielitis aguda diseminada en niños. Rev Neurol 2002; 34:163-168. [ Links ]
38. Dale RC. Acute disseminated encephalomyelitis. Semin Pediatr Infect Dis 2003, 14:90-95. [ Links ]
39. Tenenbaun S, Charoles N, Fejerman N. Acute disseminated encephalomyelitis. A long-term follow-up study of 84 pediatric patients. Neurology 2002; 59:1224-1231. [ Links ]
40. Gener B, Garaizar-Axpe C, Ruiz Espinosa C, Prats-Vinas JM. ¿Puede la encefalomielitis aguda diseminada cursar de forma diferida? Rev Neurol 2001; 32:1132-1135. [ Links ]
41. Alper G, Felice N. Towards the definition of Acute disseminated encephalitis of childhood. Curr Op Pediatr 2004; 16(6): 637-640. [ Links ]
42. Stonehouse M, Gupte G, Wassmer E, Whitehouse WP. Acute disseminated encephalomyelitis: recognition in the hands of general pediatricians. Arch Dis Child 2003; 88:122-124. [ Links ]
Autor de correspondencia: Joaquín A. Peña. MCO: 3047. P.O.Box 025233, Miami, FL 33102-5233. Telefax: + 58-61-492186. Correo electrónico: juaco949@hotmail.com

