










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInvestigación Clínica
versión impresa ISSN 0535-5133
Invest. clín vol.55 no.1 Maracaibo mar. 2014
Papel de los receptores tipo toll (TLRs) y receptores para dominios de oligomerización para la unión a nucleótidos (NLRs) en las infecciones virales.
Anyelo Durán1,2, Melchor Álvarez-Mon3 y Nereida Valero2.
1 Cátedra de Bioquímica General, Escuela de Bioanálisis, Universidad del Zulia. Maracaibo, Venezuela.
2 Sección de Virología, Instituto de Investigaciones Clínicas “Dr. Américo Negrette”, Facultad de Medicina, Universidad del Zulia. Maracaibo, Venezuela.
3 Departamento de Medicina, Universidad de Alcalá. Madrid, España.
Autor de correspondencia: Anyelo Durán. Cátedra de Bioquímica General, Escuela de Bioanálisis, Facultad de Medicina, Universidad del Zulia. Apartado Postal 23. Maracaibo 4001-A, estado Zulia, Venezuela. Telf.: +58-416-9038579, Fax: +58-261-4127005. Correo electrónico: anyeloduran@gmail.com
Resumen. Las células del sistema inmunitario (SI) son capaces de reconocer una gran variedad de microorganismos, a través de los receptores que se encuentran expresados y distribuidos a lo largo de su arquitectura celular. La interacción entre los patrones moleculares asociados a microorganismos o a daño (PMAM o PMAD) y los receptores reconocedores de patrones (RRP) presentes en las células del hospedero es un evento crítico que implica procesos intracelulares de señalización que finalizan en la expresión de mediadores tanto proinflamatorios como antivirales. Por consiguiente, de la integridad de estos receptores dependerá el buen funcionamiento de los distintos mecanismos de transducción de señal desde las membranas celulares al citoplasma y por ende, de la respuesta que el SI desencadene contra los patógenos entre ellos los agentes virales. De allí que, en esta revisión se discutirá el papel de los receptores tipo toll (TLRs) y receptores para dominios de oligomerización para la unión a nucleótidos (NLRs) en las infecciones virales, tomando como evidencia los estudios en humanos y ratones que a la fecha se conocen.
Palabras clave: TLRs, NLRs, infecciones virales, respuesta inmunitaria, señalización celular.
Role of toll-like receptors (TLRs) and nucleotide-binding oligomerization domain receptors (NLRs) in viral infections.
Abstract. The immune system (IS) cells are capable of recognizing a wide variety of microorganisms, through receptors that are expressed and distributed throughout the cell architecture. The interaction between the pathogen-associated molecular patterns or damage-associated molecular patterns (PAMPs or DAMPs) and pattern recognition receptors (PRR), present in host cells, is a critical event that involves intracellular signaling processes that end up in the expression of both, proinflammatory and antiviral mediators. Accordingly, the proper functioning of the different mechanisms of signal transduction from the cell membrane to the cytoplasm will depend on the integrity of these receptors (PRR); and therefore, the IS response triggered against pathogens including viral agents. Hence, in this review we discuss the role of toll-like receptors (TLRs) and nucleotide-binding oligomerization domain receptors (NLRs) in viral infections, using as evidence the studies in humans and mice known to date.
Keywords: TLRs, NLRs, viral infections, immune response, cell signaling.
Recibido: 18-04-2013. Aceptado: 07-11-2013
INTRODUCCIÓN
El sistema inmunitario consta de varias líneas de defensa principales. La inmunidad innata (natural o inespecífica), que carece de especificidad y de memoria, la cual constituye la primera línea de defensa del organismo, sus componentes están siempre presentes y dispuestos para actuar inmediatamente, sin requerir un tiempo de latencia para desencadenar una respuesta. Por su parte, la inmunidad adquirida (adaptativa o específica), mucho más compleja que la innata y se caracteriza por la adaptabilidad al antígeno, la especificidad y la memoria. Este tipo de inmunidad identifica péptidos específicos de patógenos procesados por células presentadoras de antígenos (CPA), las cuales, a su vez, activan las respuestas inmunitarias, mediada por células T (celular) y B (humoral). De allí, que una adecuada respuesta del sistema inmunológico dependerá de la correcta coordinación entre el sistema inmunitario innato y el adquirido (1).
Los microorganismos expresan patrones moleculares que son específicos y rápidamente diferenciables de los del hospedero; éstos incluyen virus de ARN bicatenario o doble cadena (ARNdc); dinucleótidos de citosina y guanina metilados (CpG) comunes en el ADN bacteriano, pero escasos en el ADN de vertebrados; manoproteínas de hongos; glicolípidos de las micobaterias; lipoproteínas de bacterias y parásitos; ácidos lipoteicoicos de bacterias grampositivas y lipopolisacárido (LPS) de gramnegativas. Los seres humanos a lo largo de la evolución han desarrollado receptores de reconocimiento de patrones específicos para detectar a estas moléculas asociadas a microorganismos. Dichos receptores pueden dividirse en clases: receptores secretados, endocíticos y de señalización; estos últimos son capaces de inducir la expresión de una variedad de citoquinas que subsecuentemente amplifican la respuesta inmunitaria innata y dirigen la adaptativa (1-3). Partiendo de lo anteriormente establecido, se plantea como objetivo de esta revisión describir y examinar los aspectos más importantes de los receptores reconocedores de patrones en las infecciones virales, así como analizar los trabajos realizados en esta área, en virtud de que es considerado un tema en auge donde se han centrado y siguen encaminándose muchas investigaciones y que aún falta mucho por aportar en cuanto a vías de señalización bioquímica y respuesta inmunológica se refiere. Para ello, la estructura del trabajo va desde una breve revisión general hasta el abordaje de aspectos específicos como estructura, localización, señalización y participación de estos receptores en las infecciones virales, con el fin de mostrar cómo estos receptores son capaces de interactuar con diferentes patógenos virales o no para estimular una respuesta inmunitaria específica.
GENERALIDADES
Los receptores tipo toll (TLRs, por sus siglas en inglés), son sensores de reconocimiento de membrana evolutivamente conservados, propios de la inmunidad innata que reconocen características presentes en la superficie de patógenos o que son liberados por tejido necrótico. Estas moléculas que se interpretan como “señales de peligro” forman parte de un grupo denominado patrones moleculares asociados a daño (PMAD) integrado por: los patrones moleculares asociados a microorganismos (PMAM) y las alarminas que son proteínas intracelulares liberadas por células necróticas. Los TLRs se unen a través de sus dominios, que contienen repeticiones ricas en leucina (RRL) a los PMAM, y de esa manera las células desencadenan respuestas inmunitarias, entre ellas acciones inflamatorias y antivirales a través de vías de señalización derivadas del receptor intracelular, llamado receptor Toll/IL-1 (TIR, por sus siglas en inglés de Toll/IL-1 receptor) frente a los patógenos (4-7). Los TLRs fueron llamados así por su similitud con un receptor originalmente involucrado en el desarrollo embrionario de Drosophila melanogaster descubierto en 1985 por el científico alemán Christiane Nüsslein-Volhard. En 1996, varios grupos de investigadores demostraron la importancia de estos receptores frente a los agentes micóticos en la mosca de la fruta (8). Los TLRs humanos fueron posteriormente identificados y han sido involucrados en la detección de estructuras específicas de etiología viral, microbiana, parasitaria, micótica y a señales de peligro incluso endógenas (9). Así mismo, otros de los receptores de reconocimiento de patrones (RRP), que detectan patógenos incluyen: Receptores para dominios de oligomerización para la unión a nucleótidos (NLRs), genes inducibles-I de ácido retinoico (RIG-1) también conocido como DDX58 o receptores tipo RIG1 (RLRs), y los receptores citosólicos de ADN (10, 11).
ESTRUCTURA, TIPOS Y LOCALIZACIÓN
Estructuralmente los TLRs, son similares en todo, constituido por un gran dominio extracelular (550 a 980 aminoácidos) que consiste en RRL, un dominio transmembrana y un dominio intracelular (TIR) de unos 200 aminoácidos de longitud (12). El dominio extracelular tiene la capacidad de unión al ligando mediante las RRL, siendo el responsable del reconocimiento de los diferentes PMAM, mientras que el dominio TIR media la señalización intracelular (13). Basándose en homologías de secuencia, los TLRs de vertebrados se pueden agrupar en seis subfamilias, TLR1/2/6/10, TLR3, TLR4, TLR5, TLR7/8/9 y TLR11/12/13/ 21/22/23 (14, 15). No todas las especies de vertebrados expresan todas las agrupaciones de TLRs; los seres humanos, por ejemplo, carecen de todos los miembros de la familia TLR11 (16).
Los NLRs humanos están agrupados por una familia de 22 miembros proteicos (17, 18). Formados por un dominio de reconocimiento RRL, un dominio central de oligomerización (NOD) y un dominio de señalización que puede contener dominios de reclutamiento y activación de caspasas (CARD), dominios de pirina o dominios inhibidores de apoptosis que participan en la activación de diferentes vías de señalización (19, 20). Los miembros de la familia NLRs están agrupados en al menos cinco subfamilias distinguidos por sus estructuras amino terminales. Éstas incluyen los NLRA, que contienen un dominio de transactivación, los NLRB los cuales contienen unas proteínas inhibidoras de la apoptosis de baculovirus (baculovirus IAP, BIR; por sus siglas en inglés), los NLRC, que contienen un dominio de reclutamiento de caspasas (CARD; por sus siglas inglés), los NLRP, contienen un dominio pirina conocido también como dominio proteico y los NLRX con un dominio desconocido hasta la fecha. Hasta ahora hay 23 genes para NLRs humanos y 34 han sido identificados en el modelo múrido, la función fisiológica de la mayoría de los NLRs es pobremente entendida (21, 22).
Los TLRs son expresados en diferentes tipos celulares, principalmente en las del sistema inmunitario tales como células dendríticas, macrófagos, neutrófilos y linfocitos, además de las células endoteliales y epiteliales. Acorde a la localización, los TLRs han sido dividido en dos categorías: los que se localizan en la superficie de la membrana celular (TLR1/2/4/5/6/10) y los que encuentran principalmente en la membrana de los endosomas (TLR3/7/8/9) (13, 23-25). Los NLRs se localizan en el citosol celular, y desde allí reconocen bacterias intracelulares, productos microbianos y ácidos nucleicos de virus, así como otras señales de peligro intracelular, iniciando así, las vías de defensa del hospedero a través de la activación de caspasas inflamatorias, y la respuesta generada por la traslocación del factor nuclear kappa B (NF-kB, por sus siglas en inglés) (26, 27).
VÍAS DE SEÑALIZACIÓN ACTIVADAS POR LOS TLRs Y NLRs
La interacción entre los TLRs y los PMAD dan origen a una secuencia de eventos intracelulares que culminan con la expresión de genes relacionados con la inflamación, dependiendo de la naturaleza de la señal de peligro y del o los TLRs implicados involucran la activación de diferentes vías de señalización. El funcionamiento de estas rutas puede ser dependiente o independiente de la proteína acopladora conocida como factor de diferenciación mieloide 88 (MyD88) (11).
La traslocación de algunos TLRs desde el retículo endoplasmático (RE) a la membrana del endosoma es mediada por algunas proteínas residentes ubicadas en el RE tales como; UNC93B1 (para el TLR3/7/8/9), proteína asociada al TLR4 (PRAT4A) para TLR1/2/4 desde el RE a la membrana plasmática (MP) y del TLR7/9 desde el RE al endosoma, no siendo necesaria la presencia de estas proteínas chaperonas para el tráfico del TLR3 (23). Una vez en el endosoma, los TLR3/7/8/9 están sujetos a procesos de escisión proteolítica, los cuales son requeridos para la unión del ligando y la consiguiente señalización (23, 24, 28) (Fig. 1). Para algunos TLRs, la unión del ligando se ve facilitada por correceptores proteicos, que incluyen al grupo de diferenciación 14 (CD14, por sus siglas en inglés) y la proteína asociada al dominio extracelular del TLR4 (MD2, por sus siglas en inglés). Tras el acoplamiento del ligando, con el dominio citoplasmático de los TLRs se reclutan los adaptadores de señalización MyD88, la proteína asociada a TIR (TIRAP) y molécula adaptadora que contiene TIR (TRAM) y/o TRIF eventos estos, que ocurren en el dominio TIR de los TLRs. Dependiendo de la naturaleza del adaptador que se emplee, éste se une a la cinasa asociada al receptor de IL-1 (IRAK4, IRAK1, IRAK2, TBK1 y IKKe), las cuales, a su vez, se unen al factor asociado al receptor del TNF-6 (TRAF-6) para activarlo y estimular a TAK1; esta cinasa pone en marcha la señalización por la proteína cinasa MAKK, quien fosforila a otras cinasas como JNK para generar la activación y translocación de factores nucleares como el PA-1 y el NF-kB, con la consecuente transcripción de genes que codifican para citoquinas proinflamatorias. Seguidamente la cinasa IRAK-1, es capaz de activar el factor regulador de interferón 7 (IRF-7, por sus siglas en inglés) induciendo la transcripción de los genes que codifican para los IFN tipo I (3, 29). La señalización del TLR4 a través de la proteína adaptadora TRIF conduce a la activación de TRAF6, que modifica a TAK1, permitiendo que el complejo TRAF6-TAK1 active la MAKK, promoviendo la producción de citoquinas inflamatorias. Alternativamente, la señalización del TLR4 a través de MyD88 conduce a la activación de TRAF3, que estimula a TBK1, activando al factor regulador de interferón 3 (IRF-3, por sus siglas en inglés) quien promueve a nivel nuclear la transcripción de genes conducentes a la producción de los IFN tipo I (30) (Fig. 2). La señalización de los TLRs puede terminar mediante reguladores negativos como: IRAK-M y TOLLIP, que antagonizan la activación de IRAK1; FADD que antagoniza tanto a MyD88 como a IRAK1. Mientras que A20 bloquea a TRAF6 y a IKK (3, 31). Las fallas en la cascada de señalización de los TLRs se han asociado con enfermedades graves para los humanos. Los pacientes con deficiencias hereditarias de MyD88, IRAK4, UNC93B1, o del TLR3 muestran un patrón incrementado de susceptibilidad a infecciones recurrentes de etiología viral y bacteriana (32). La activación crónica del TLR7/9 en las células B auto reactivas, se ha relacionado en mayor grado con la aparición de enfermedades autoinmunes sistémicas (33). Además, la activación de mutaciones oncogénicas en MyD88 ocurre con frecuencia en un subtipo de células B activadas como el linfoma difuso de células B grandes y en otras malignidades de las células B (34). Varios agonistas del TLR7 y TLR9 se están probando en los ensayos clínicos como adyuvantes para aumentar las respuestas antitumorales en los pacientes con cáncer (35).

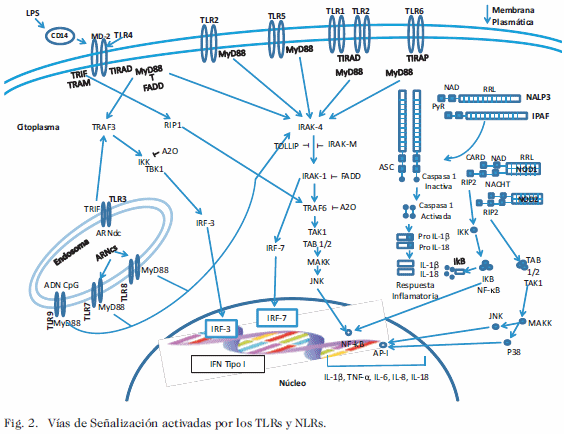
La señalización mediada por los NLRs entre sus representantes el NOD1 y NOD2 se ha descrito que éstos se unen a través de sus dominios de reclutamiento y activación de caspasas (CARD) a la proteína cinasa RIP2 (RICK), quien recluta a IKK, TAB1/2 y TAK1 con la consecuente activación del complejo MAKK quien fosforila a la JNK y P38 los cuales se traslocan a nivel nuclear afectando la expresión de genes a través del NFkB y del AP-1 promoviendo así la producción de citoquinas proinflamatorias (IL-1, TNF-a, IL-4, IL-12, IL-6, IL-8, IL-18); mientras que otros receptores como el IPAF y el complejo multiproteico de caspasa 3 (NALP1-3) parecen tener control postraduccional del procesamiento y secreción de IL-1b e IL-18 por un mecanismo dependiente de caspasa 1 y la proteína asociada a la apoptosis portadora de dominio CARD (ASC) denominado inflamasoma, generando así una respuesta inflamatoria (19, 20) (Fig. 2).
TLRs Y NLRs EN LAS INFECCIONES VIRALES
Son varios los miembros de la familia TLRs a los que se les han atribuido implicaciones en respuestas a las infecciones virales entre los cuales tenemos: TLR1/2/3/4/ 6/7/8/9 (36) y de los NLRs al NOD2 (37) y NLRP3 (38, 39). La contribución de cada uno de estos receptores en las infecciones virales se discute a continuación. En particular, la evidencia emergente que sugiere un papel para los TLRs y NLRs humanos en la respuesta antiviral.
SUBFAMILIA 1 (TLR1/2/6/10)
La respuesta inmunitaria desencadenada por las células ante la activación del TLR2 ha sido documentado para un amplio número de virus de ADN tales como Citomegalovirus (CMV), Herpes Simple (VHS), virus de Epstein-Barr (VEB), virus Vacuna (VV) y virus de ARN tales como el virus de la Coriomeningitis Linfocítica (LCMV), Hepatitis C (VHC) y el Virus Sincicial Respiratorio (VSR) (40-46). El TLR2 desencadena respuestas antivirales mediadas por diferentes tipos celulares. Por ejemplo, el LCMV en las células gliales del sistema nervioso central (SNC) (41), VEB en monocitos (42) y VHS en las células microgliales (47). En fibroblastos humanos un heterodímero del TLR1/2 reconoce las proteínas de la envoltura de CMV y desencadena respuestas inmunitarias conducentes a la secreción de citoquinas proinflamatorias (43-46, 48). En leucocitos se demostró el papel del TLR2 en la respuesta antiviral contra VSR. Se ha demostrado que el heterodímero TLR2/6 es importante en la respuesta de citoquinas a VSR y en el control de la replicación viral in vivo. Además, la migración de neutrófilos y células dendríticas (CDs) activadas en el pulmón, son eventos dependientes de la interacción del TLR2 con el VSR (44).
El papel del TLR2 en la detección de infección por VV fue el primer reporte informado en un estudio utilizando CDs derivadas de médula ósea, donde la respuesta de citoquinas proinflamatorias ha demostrado ser dependiente del TLR2, mientras que la respuesta de IFN tipo I ha demostrado ser independiente para este receptor (45). Esto de acuerdo a estudios que han demostrado que la señalización del TLR2 dependiente de MyD88 no estuvo involucrada con la producción de IFN tipo I. Sin embargo, este hallazgo difiere a los reportados por otros autores donde se ha conseguido que el TLR2, en monocitos especializados, los llamados monocitos inflamatorios (Ly6Chi), pueden inducir IFN tipo I tras el estímulo de partículas virales como el VV y CMV (49).
Investigadores han sugerido que algunos virus pueden utilizar el TLR2 para su propio beneficio. No obstante, hay casos de infecciones virales donde este receptor puede no ser utilizado. En este estudio, mediante el uso de anticuerpos de bloqueo demostraron que la proteína del core del VHC activaba la expresión de IL-10 y TNF-a a través del TLR2 en monocitos humanos. Ambas citoquinas causan una reducción en la liberación de IFN-a e incremento de la apoptosis por parte de las células dendríticas plasmocitoides (CDsp) (50).
Otros autores han señalado que el VHC es reconocido por el hospedero a través del heterodímero TLR2/TLR6, el cual reconoce a la proteína de core del virus, así como a la proteína no estructural 3 (NS3), pudiendo además este ligando viral interactuar con el TLR3, 7 y 8 (51, 52). El reconocimiento del VHC por el TLR2 activa a monocitos y macrófagos humanos los cuales secretan principalmente IL-10 y TNF-a, pero inhiben la función de diferenciación de macrófagos a CDs (51). Por consiguiente, los pacientes con hepatitis crónica presentan aumento en la expresión del TLR2 en las células mononucleares de sangre periférica (CMSP), correlacionada con el aumento circulante de TNF-a lo que favorece la actividad inflamatoria y de necrosis a nivel hepático (53, 54). La activación de las CMSP por medio del TLR2/4 en pacientes con hepatitis C crónica inducidas por el ligando viral resultaron en niveles incrementados de IL-6 en comparación con individuos sanos, probablemente desencadenado por el TLR2 (55).
Por otro lado, Brown y col. (56) demostraron que el polimorfismo R753Q en el TLR2 perjudica el reconocimiento por parte de este receptor a la proteína del core del VHC y a la NS3, lo que aumenta el riesgo de fracaso al trasplante hepático en los pacientes que cursan con hepatitis crónica y a los cuales se les ha detectado esta mutación. Este es un ejemplo del contraste de papeles que el TLR2 puede jugar durante los procesos infecciosos en los seres humanos.
El virus dengue (DV) causa una de las infecciones virales más comunes en países tropicales y sub-tropicales, transmitida por mosquitos, amenazando la salud humana en más de 100 países alrededor del mundo (57-59). Ante esta infección la producción de IFN tipo I y de citoquinas es crucial para una adecuada respuesta inmunitaria contra este agente, de manera que estudios anteriores refieren que el tipo 2 (DV-2) induce niveles bajos del IRF-3 con la consecuente menor activación del NF-kB, lo que conduce a una menor producción de IFN-b en la fase temprana de la infección (60-62). Chang y col. (63) determinaron que la infección por DV no sólo impide la activación de IFN tipo I, sino también la producción de citoquinas desencadenada por la señalización de los TLRs. En este estudio se reportaron bajos niveles de IFN-b así como de IL-10, IL-12 y TNF-a. Por otra parte, la infección producida por DV-2 fue reprimida en parte por citoquinas como consecuencia de la señalización del TLR. Igualmente se encontró que la activación de NF-kB activada por ligandos de TLR fue bloqueada por DV-2. A su vez, la señal extracelular reguladora de cinasa (ERK) fue suprimida por el virus, concluyendo que el DV-2 por sí mismo es un inductor débil de IFN tipo I y de citoquinas, además este virus puede bloquear el TLR impidiendo la translocación del NF-kB y su activación con la consiguiente disminución en la producción de citoquinas.
SUBFAMILIA 2 (TLR3)
El TLR3, es expresado en los endosomas de células dendríticas mieloides (CDsm), y al igual que otros TLRs es capaz de reconocer ácidos nucleicos. Sin embargo, en fibroblastos y células epiteliales, el TLR3 es expresado en la superficie celular (64). También se ha reportado su expresión abundante en cerebro, especialmente en neuronas, astrocitos y microglia (65-67). El TLR3, ha demostrado ser un receptor de reconocimiento de algunos virus de ARNdc y también puede detectar virus de ADN que generan durante su ciclo de vida ARNdc (68).
En estudios experimentales con ratones deficientes de TLR3 a los cuales se expusieron a diferentes agentes virales, entre ellos el LCMV, virus de la Estomatitis Vesicular (VSV), CMV y Reovirus, la respuesta inmunitaria desencadenada producto de la activación de este receptor generó un efecto benéfico para el animal desde el punto de vista inmunológico (64, 69). Contrario a otros estudios de infección viral, donde el TLR3 mostró mediar inmunidad perjudicial para el hospedador. Utilizando ratones deficientes en TLR3, éstos mostraron una menor resistencia a la infección a un número considerable de virus, incluyendo Punta Toro, VV e Influenza, se cree que es debido a que el TLR3 regula una sobreproducción de mediadores inflamatorios (70-72). Estos son ejemplos donde la inmunidad mediada por el TLR3 favorece al virus y puede ser visto como un mecanismo de evasión viral (73).
El estudio de la relación del TLR3 con el virus del Nilo Occidental (VNO) arrojó que el TLR3 contribuye a la letalidad por este virus al promover la inflamación periférica que conduce a la ruptura de la barrera hematoencefálica (BHE), lo que resulta en un aumento de la carga viral en el cerebro. Por lo tanto, los ratones que carecen de TLR3 eran más resistentes a la letalidad por el VNO en comparación con ratones que presentan este receptor (74). Sin embargo, un estudio posterior reportó resultados opuestos, donde el TLR3 demostró tener un papel protector contra la infección por el VNO por el cual la ausencia de TLR3 disminuye la supervivencia de los ratones en respuesta a la presencia de VNO y la carga viral fue mayor en el cerebro (75). Aunque los datos relativos a TLR3 y VNO son controvertidos, otra evidencia en apoyo al papel positivo del TLR3 en la inmunidad protectora contra el VNO proviene de las observaciones de que la proteína NS1 y la proteína de la envoltura del VNO inhibe la señalización del TLR3 al inhibir al receptor interactuante con RIP1, una proteína necesaria para la señalización en la vía de traducción de señal del TLR3 (76, 77).
Estudios utilizando ratones knock-out han reportado la implicación que tienen los TLRs en la respuesta antiviral con respecto a los datos que se tienen de los TLRs humanos la cual es más limitada. Sin embargo, el papel del TLR3 humano para proporcionar un rol protector contra la encefalitis por VHS fue descrito en dos niños que albergaban una mutación heterocigótica en el TLR3. Esta mutación (C a T) dio como resultado la sustitución de una prolina por una serina en el aminoácido 554 del dominio extracelular que cuenta con 20 RRL. Esta mutación se comportó de forma autosómica dominante con una predisposición específica a la encefalitis por VHS, mientras que la inmunidad a otros virus no se afectó. Este es el primer trabajo que demostró de manera concluyente una relación entre la inmunidad mediada por TLR3 hacia agentes virales, particularmente a nivel del SNC (78). Además, el ARN liberado a partir de células infectadas con VEB fue capaz de activar el TLR3 humano. De manera que los pacientes crónicamente infectados con este virus desencadenaron la señalización del TLR3 que activa a linfocitos y células mononucleares periféricas (79).
Liang y col. (80) presentaron un trabajo sobre activación del TLR3 por medio del DV-2 e inducción de IFN-b en cultivos de células de hepatoma. Este trabajo tuvo como objetivo investigar el papel de la respuesta inmunitaria innata en células infectadas por DV, demostraron que la activación del TLR3 tuvo un marcado efecto antiviral, en tanto que el pretratamiento de otros ligandos de TLRs (incluyendo TLR1/2, TLR2/6, TLR4, TLR5 y TLR7/8) no mostró un efecto significativo.
En la infección por virus de Influenza se ha evaluado la acción antivírica de agonistas ácido-base para la activación de los TLRs. Se ha observado que el TLR3 expresado en las CDs del epitelio respiratorio y macrófagos desempeña un papel central en la mediación de la respuesta inflamatoria de la inmunidad innata frente a las infecciones virales. Los virus de influenza pueden inhibir la capacidad de la célula hospedera para producir IFN, así como suprimir los mecanismos de defensa anti-viral del sistema inmunitario. Se ha evidenciado en ratones que la administración intranasal de ácido poliinosínico-policitidílico (poli I:C), y de poli I:C encapsulado en liposomas, que son moléculas agonistas del TLR3 e inductoras de IFN y linfocitos citolíticos, confiere un alto grado de protección frente al virus de influenza A/H5N1. La duración de este efecto protector persistió hasta 3 semanas para el poli I:C encapsulado en liposomas y 2 semanas para el poli I:C. De forma similar, el tratamiento previo en ratones con CpG (agonista del TLR9) confería una completa protección ante la infección del virus de Influenza A (81).
Los macrófagos son CPA, que expresan TLRs y NLRs quienes tienen un papel clave en la respuesta inmunitaria contra los virus. Los coronavirus (CoVs) son virus de ARN monocatenario, de sentido positivo que causan infecciones agudas y crónicas, pudiendo infectar a los macrófagos, siendo estas células suficientemente permisivas a la infección. El estudio de interacción entre CoVs y los RRP se encuentra en pleno desarrollo (82, 83). En este contexto, un grupo de investigadores analizaron el efecto de la activación del TLR2/3/4/7 mediante el empleo de ligandos específicos en la línea celular de macrófagos J774A.1 susceptibles a la infección con coronavirus múrido (virus de la hepatitis de ratón [VHR]). La estimulación de los TLR2/4/7 no afectó la producción de VHR. En contraste, la pre-estimulación de TLR3 con poli I:C limitó la infección por VHR a través de la inducción de IFN-b en los macrófagos. Este estudio demostró la activación del TLR3 con el ligando sintético poli I:C el cual media una inmunidad antiviral incrementada en la cepa neurotrópica VHR-A59 o suprime la producción de virus en la cepa neurotrópica, VHR-JHM, y la cepa hepatovirulenta VHR-3 en macrófagos infectados (83).
SUBFAMILIA 3 (TLR4)
La evidencia inicial que TLR4 podría ser un RRP anti-viral fue demostrada en un estudio que muestra que la proteína de fusión (F) del VSR estimula la producción de citoquinas a través del TLR4 (84). Estudios posteriores de infección por VSR en ratones knock-out para TLR4 mostraron una actividad reducida de la función celular de las NK, así como disminución de la expresión de IL-12 y eliminación alterada del virus en comparación con el grupo de ratones controles (85). En cuanto a la infección por VV, los ratones que carecen de TLR4 mostraron una mayor replicación viral y mortalidad en comparación con los ratones controles tras la infección respiratoria. Se cree que el TLR4 está implicado en mediar una inmunidad protectora contra VV debido a que todavía no se ha detectado un ligando viral para TLR4 en ratones (86). En los macrófagos se demostró que VSV activa la cinasa PI3 a través del TLR4, dando lugar a la expresión de IFN tipo I confiriendo inmunidad anti-viral (87). En contraste, otros sugieren que el TLR4 contribuye de manera perjudicial durante la respuesta inmunitaria hacia infecciones virales como por ejemplo: El reconocimiento de la glicoproteína del virus Ébola por el TLR4 humano conduce a la producción de citoquinas proinflamatorias y se cree puede mediar la inmunopatogénesis viral (88).
Suh y col. (89) en un estudio en microglia humana demostraron que la activación del TLR4 puede inhibir una vía de replicación del virus de la inmunodeficiencia humana (VIH) que es requerida por el IRF-3. Estudios relacionados, han encontrado que polimorfismos en el TLR4, pueden influir al incrementar la carga viral en individuos infectados por VIH (90). La importancia del TLR4 humano de lactantes con infección por VSR sugiere, que la presencia de mutaciones en el TLR4 Asp299Gly o Thr399Ile se asoció con un mayor riesgo de bronquiolitis grave por este virus (91). Mientras que en pacientes con hepatitis B crónica, se demostró una sobreexpresión de los TLR4 y TLR2 en monocitos, modulando las actividades de las células T reguladoras (Treg) que pueden contribuir a la inmunotolerancia (92).
En cuanto al efecto de la activación del TLR4 sobre la replicación del DV, sólo unos pocos estudios han sido reportados. Un grupo encontró que el LPS es capaz de inhibir la infección por DV en monocitos/macrófagos mediante el bloqueo de la entrada viral (93), contrario a ello se ha demostrado que el pretratamiento con LPS no redujo significativamente la replicación del DV en las células HepG2 (94). Curiosamente, se encontró que el DV puede contrarrestar la activación de la vía del TLR, lo que implica que la señalización del TLR puede tener algunas funciones en el bloqueo de la infección por DV (95).
SUBFAMILIA 5 (TLR7/8/9)
Los TLR7 y TLR8 están relacionados filogenética y funcionalmente. Y han sido identificados como importantes sensores para el reconocimiento de genomas virales de ARN de cadena sencilla (ARNcs), como: Influenza, VSV y VIH (96, 97). Aunque alguna vez se pensó que el TLR8 en ratones no era funcional (98), ahora se sabe que el TLR8 múrido se puede activar en algunas circunstancias (99). La contribución de estos receptores a la inmunidad proviene del hecho que el TLR7 es expresado en las CDsp y el TLR8 en las CDsm. Tanto el ratón como el humano expresan TLR7 en las CDsp las cuales son las responsable de la producción de altos niveles de IFN tipo I, importante en la inducción de respuestas del patrón Th1, y en el cambio de clase de inmunoglobulinas por las células B (100). Además, las células B que expresan TLR7 responden a virus de ARNcs por la activación de moléculas co-estimuladoras y la producción de citoquinas (97). Una contribución importante del TLR7 a la inmunidad contra el VNO se demostró en ratones knock-out, deficientes de TLR7 y MyD88 en los que se observó un incremento de la replicación viral y mayor mortalidad comparada con el grupo control. Así como también un número reducido de macrófagos, células T CD4+ y CD8+ a la infección por VNO en el cerebro de los ratones infectados, que ha demostrado ser dependiente del incremento de la IL-23 (101, 102). Sin embargo, otro estudio pone en duda la participación del TLR7 en la inmunidad contra el VNO, dado que la susceptibilidad al virus frente al TLR7 no difirió en ratones controles que no tenían el gen del TLR (TLR-/-) ratones knock-out y ratones que si poseían el gen para el receptor tipo toll (TLR+/+) después de la infección intradérmica. En ratones (TLR+/+) hubo reducción del número de CD11c+ presentes en las células de Langerhans en la epidermis después de la infección intradérmica, un efecto no observado en los ratones deficientes de TLR7. Estos investigadores concluyeron que, después de la infección cutánea, la respuesta inmunitaria mediada por TLR7 contribuye a la patogénesis viral mediante la diseminación del VNO por la piel a otros órganos para iniciar una infección sistémica. Se ha propuesto que este proceso podría reducir la inmunidad protectora mediada por TLR7 durante la etapa sistémica de la infección. El caso del VNO con el TLR7 ilustra que es preciso definir el papel exacto de los TLRs en las infecciones virales, lo cual resulta difícil y depende de factores tales como la dosis viral, el número de pases y la vía de administración del virus (103).
Se ha demostrado la importancia del TLR7 humano en la patogénesis del VIH. Meier y col. (104) en su estudio, reportaron que el TLR7 puede ser un factor crucial para explicar por qué las mujeres infectadas tienen cargas virales más bajas a principios de la infección, pero el progreso del síndrome de inmunodeficiencia adquirida (SIDA) es más violento que en los hombres. Se encontró que las CDsp de las mujeres producían niveles significativamente más altos de IFN-a en respuesta al VIH-1 como resultado de una mayor estimulación del TLR7 en comparación con las CDsp de los hombres, después de ajustar la carga viral. Además, también había mayor número de linfocitos T CD8+ activados en las mujeres con infección crónica por el VIH-1. Se piensa que este aumento del nivel de activación frente al VIH en las mujeres puede conducir a una mayor progresión de la enfermedad. Estos datos sugieren que la inhibición de la vía de TLR7 en las CDsp podría representar un nuevo enfoque para tratar infecciones por el VIH-1. Curiosamente, un polimorfismo en el TLR7, Gln11Leu, ha sido asociado con cargas virales más altas y una progresión más acelerada de la enfermedad en personas infectadas con VIH (105).
La respuesta inmunitaria inducida por la expresión del TLR9 suscita activación en un número variado de células, tales como neutrófilos, monocitos y células T CD4+, así como también de un número de células no inmunitarias tales como células epiteliales las cuales también expresan TLR9. En contraste, el TLR9 se expresa constitutivamente en las células B y CDsp (106). El TLR9 puede detectar CpG presentes en el ADN microbiano de allí que este receptor participa en la respuesta inmunitaria a virus de ADN. La respuesta antiviral que depende del TLR9 se debe en gran parte a la producción de grandes cantidades de IFN tipo I mediada por las CDsp. Estas acciones se producen en respuesta a la infección con CMV (107), VHS-1 (108), adenovirus (109) y virus de la viruela (110).
En relación al TLR9 humano, las investigaciones sugieren que éste es importante en la infección por VIH. Así se ha reportado reducción en la expresión del TLR9 en las células B de personas infectadas por este virus, lo que lleva a un deterioro de estas células en respuesta al agonista para el TLR9 (111). Además, se ha descrito que los CpG aumentan la respuesta de las células B en individuos infectados por VIH (112). También se ha demostrado que la glicoproteína (gp120) del virus suprime la activación de CDsp humanas después de la estimulación por el TLR9 (113). Igualmente ha sido documentado que el polimorfismo en el TLR9 afecta el resultado clínico de la infección por VIH-1, que conduce a una progresión de la enfermedad de forma más rápida. Dado que el VIH es un virus de ARN no se conoce si el TLR9 directamente interactúa con el virus, o si la respuesta inmunitaria al VIH mediada por TLR9 se produce como una consecuencia secundaria (114).
Otros ejemplos de modulación del TLR9 humano por infecciones virales son los pacientes con infección crónica por VHB, donde la expresión de este receptor se reduce en las CDsp (115), al igual que en la infección causada por el VHC, lo que sugiere que el TLR9 podría ser importante durante estas infecciones (116). Además, en las infecciones causadas por el VSR y sarampión, el TLR9 y TLR7 median la producción de IFN tipo I la cual es inhibida en las CDsp de humanos (117). Aunque se ha sugerido que el TLR9 podría mediar una respuesta perjudicial durante la infección bacteriana (118) y CpG en el ADN lleva a la activación y la replicación del VIH en células esplénicas de ratón (119), no existen hasta ahora estudios que sostengan con firmeza que el TLR9 promueve inmunidad perjudicial durante la infección viral, como se ha reportado para el TLR3. Además, no hay trabajos que indiquen que el TLR9 es utilizado por el virus como un medio de diseminación sistémica. En contraste, se ha informado de que TLR7 y TLR8 de neutrófilos humanos podría contribuir a la patogénesis de la infección por los virus de la influenza (120).
NOD2
Sabbah y col. (37), en su trabajo sobre la activación de la respuesta inmunitaria antiviral por NOD2, demostraron que el NOD2 funciona como un RRP citoplasmático frente a agentes virales que media la activación del IRF-3 con la consiguiente producción de IFN. Después del reconocimiento del genoma del VSR, el NOD2 utiliza las proteínas adaptadoras de señalización mitocondrial antiviral (Mavs) para activar el IRF-3. Ratones deficientes de NOD2 no producen IFN de manera eficiente, y han mostrado una mayor susceptibilidad al VSR y consecuentemente una mayor patogénesis. Así, la función de NOD2 como un RRP destaca como mecanismo antiviral de defensa del hospedador.
NLRP3
A través de estudios se ha sugerido que los ARNdc y poli I:C activan el inflamasoma mediante una vía dependiente de NALP3; lo que no está muy claro es si el NALP3 reconoce directamente al ácido nucleico en el citoplasma (38). NALP3 es también crítico para la producción de IL-1b como respuesta a la infección por adenovirus. Aunque los adenovirus son virus de ADN, la introducción de ADN de doble cadena (ADNdc) en el interior celular ha sido asociado con la producción de IL-1b la cual obedece a la activación de manera independiente de NALP3, lo que sugiere que el adenovirus induce la producción de IL-1b y ésta no es mediada por el reconocimiento de ADN genómico. Sin embargo, el adaptador asociado a apoptosis o la proteína de unión a CARD (ASC) y la caspasa-1 son esenciales para la producción de IL-1b inducida por ADNdc por lo que se sugiere que uno de los miembros no conocido de la familia NALP su función es servir de sensor para el reconocimiento del ADN citoplasmático (39).
PERSPECTIVAS FUTURAS
La inmunidad innata desempeña un papel importante en la iniciación de una respuesta inmunitaria efectiva frente a los diferentes antígenos a los cuales estamos expuestos o con los que interactuamos. Se sabe que los RRP reconocen microorganismos patógenos y no patógenos, así como componentes generados por estrés celular y daño no microbiano. Por ello, es importante continuar las investigaciones que permitan comprender su participación en el contexto específico de los microorganismos que causan enfermedades con la finalidad de alcanzar un mejor entendimiento en los mecanismos de respuesta inmunológica lo cual será de utilidad, para el futuro desarrollo de terapias innovadoras en el tratamiento de enfermedades infecciosas o no infecciosas.
Hasta la fecha se han logrado grandes avances en la comprensión de la respuesta del sistema inmunitario mediada por los TLRs y NLRs después de su interacción con diferentes virus. Sin embargo, no todos los eventos de señalización y proteínas necesarias están plenamente detallados, así como sus diferentes formas de regulación. También está claro que el resultado de una interacción virus-TLRs/NLRs es complejo y depende del receptor en particular y del virus en cuestión así como, las especies hospedadoras. La respuesta inmunitaria para al menos algunas infecciones virales requiere la contribución de muchos RRP haciendo el proceso de respuesta inmunológica algo complejo para la infección natural sobre todo porque para muchos agentes, estas señales de integración se desconocen.
Se han encontrado diferencias importantes de señalización por los TLRs en ratones y humanos, por lo tanto, la manipulación terapéutica de los TLRs requerirá una comprensión más completa en la especie humana. Debido a que los TLRs pueden mediar respuestas inmunitarias desfavorables después de la infección viral, futuros estudios deberán aportar o sugerir blancos de drogas para los TLRs, o para algún o algunos elementos implicados en la(s) vía(s) de señalización que puedan ser moduladas con el fin de mejorar el resultado a diferentes infecciones virales. Es posible que algunos de estos medicamentos resulten ser la base de inhibidores virales para la señalización de los TLRs y NLRs. Es necesario dirigir la atención al estudio del polimorfismo génico en la familia de los TLRs y de los NLRs para así poder comprender la posible vinculación con los eventos inflamatorios, de desregulación y/o ausencia e incremento de la respuesta inmunitaria del hospedador hacia el patógeno.
CONCLUSIÓN
Los TLRs y los NLRs son proteínas transmembranales y citosólicas respectivamente, altamente conservadas a través de la evolución, presentes en una amplia gama de células de animales invertebrados y vertebrados. Estos receptores desempeñan funciones básicas frente a la respuesta inmunitaria innata detectando la presencia de microorganismos, y generando de manera rápida una respuesta frente al antígeno que en fases posteriores estimulan el desarrollo de una respuesta adaptativa duradera y más eficaz. Nuestra comprensión conceptual de cómo los TLRs y los NLRs reconocen los virus se ha expandido de manera significativa durante los últimos años. Recientes descubrimientos demuestran un papel importante de estos receptores en el reconocimiento de células infectadas por virus que tienen tropismos por diferentes líneas celulares tanto en el modelo múrido, como en humanos. Este reconocimiento viral por parte de los TLRs conduce a la producción de IFN de tipo I. De allí que, el papel de esta citoquina en la generación de una respuesta antiviral es crucial, y la inhibición de estas vías de señalización pareciera ser un común que utilizan muchos virus como un mecanismo de evasión viral al sistema inmunitario. Debido a estas funciones los TLRs y los NLRs, han generado gran expectativa, por lo cual se viene estudiando las alteraciones de su expresión relacionados a la susceptibilidad y resistencia frente a enfermedades, así como la producción de fármacos que promuevan su estimulación en el control de enfermedades inmunitarias y cáncer, y muy especialmente en aquellas infecciones virales que no cuentan con mecanismos de prevención por inmunizaciones.
AGRADECIMIENTOS
Este trabajo fue apoyado por los Programas de Investigación financiados tanto por el Consejo de Desarrollo Científico, Humanístico y Tecnológico de la Universidad del Zulia (CONDES CC-0393-12; CC-0291 -13) como del Subproyecto de Misión Ciencia N° 2008000911-1. Le damos las gracias al Dr. Jesús Mosquera por la evaluación crítica a esta revisión y al Univ. Luis Gallardo por la elaboración de las figuras.
REFERENCIAS
1. Abbas AK, Lichtman AH, Pillai S. Sección I: Introducción al sistema inmunitario: Propiedades generales de las respuestas inmunitarias e inmunidad innata en: Inmunología celular y molecular; 2008, p 3-46. Sección IV: Mecanismos efectores de la respuesta inmunitaria: Citocinas en: Inmunología celular y molecular. Elsiever España, Madrid 6ª Edición. p 267-301. [ Links ]
2. Janeway J, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002; 20: 197-216. [ Links ]
3. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11:373-384. [ Links ]
4. Bell JK, Mullen GE, Leifer CA, Mazzoni A, Davies DR, Segal DM. Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol 2003; 24: 528-533. [ Links ]
5. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol 2003; 21:335-376. [ Links ]
6. Bianchi ME. DAMPs, PAMPs and alarmins: All we need to know about danger. J Leukoc Biol 2007; 81(1):1-5. [ Links ]
7. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 2012; 4: pii: a006049. [doi: 10.1101/cshperspect.a006049]. Available from: URL: http://cshperspectives.cshlp.org/content/4/3/a006049.
8. Lemaitre B, Nicolas E, Michaut L, Reichhart JM and Hoffmann JA. The dorso ventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996; 86:973-983. [ Links ]
9. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 2009; 21:317-337. [ Links ]
10. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458:514-518.
11. Yanai H, Savitsky D, Tamura T, Taniguchi T. Regulation of the cytosolic DNA-sensing system in innate immunity: a current view. Curr Opin Immunol 2009; 21:17-22.
12. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol 2001; 1:135-145.
13. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783-801.
14. Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, Kuroki Y. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC. Genomics 2007; 8:124. [doi: 10.1186/1471-2164-8-124]. Available from: URL: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1899181.
15. Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood L, Aderem A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. U. S. A. 2005; 102:9577-9582.
16. Botosa I, Segalb D, Daviesa D. The Structural Biology of Toll-Like Receptors, Structure 2011; 19(4): 447-459.
17. Faustin B, Reed JC. Sunburned skin activates inflammasomes. Trends Cell Biol 2008; 18: 4-8.
18. Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE. The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol 2007; 29: 289-301.
19. Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 2006; 6:9-19.
20. Inohara N, Chamaillard M, McDonald C, Núñez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem 2005; 74:355-383.
21. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA. The NLR gene family: a standard nomenclature. Immunity 2008; 28(3):285-287.
22. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140: 821-832.
23. Blasius AL, Beutler B. Intracellular Toll-like receptors. Immunity 2010; 32: 305-315.
24. McGettrick AF, O’Neill LA. Localisation and trafficking of Toll-like receptors: An important mode of regulation. Curr Opin Immunol 2010; 22: 20-27.
25. Lim KH, Staudt LM. Toll-Like Receptor Signaling. Cold Spring Harb Perspect Biol, 2013; 5:a011247. (doi: 10.1101/csh perspect.a011247). Available from: URL: http://www.ncbi.nlm.nih.gov/pubmed/23284045.
26. Zhang X, Mosser DM. Macrophage activation by endogenous danger signals. J Pathol 2008; 214:161-178.
27. Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science 2010; 327: 286-290.
28. Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: Regulation through compartmentalization. Nat Rev Immunol 2009; 9:535-542.
29. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol 2012; 4 (11): pii: a011254. (doi: 10.1101/cshperspect. a011254). Available from: URL: http://cshperspectives.cshlp.org/content/4/11/a011254.
30. Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 2011; 11: 457-468.
31. Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: Critical regulators of innate immune signalling. Biochem Pharmacol 2010; 80:1981-1991.
32. Casanova JL, Abel L, Quintana-Murci L. Human TLRs and IL-1Rs in host defense: Natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol 2011; 29: 447-491.
33. Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol 2011; 23: 106-112.
34. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470: 115-119.
35. Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: Emerging therapeutics? Nat Rev Drug Discov 2010; 9: 293-307.
36. Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev 2009; 227: 75-86.
37. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y, Bose S. Activation of innate immune antiviral responses by Nod2. Nature Immunology 2009; 10: 1073-1080.
38. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Núñez G. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and doublestranded RNA. J Biol Chem 2006; 281: 36560-36568.
39. Muruve DA, Pétrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008; 452:103-107.
40. Sorensen LN, Reinert LS, Malmgaard L, Bartholdy C, Thomsen AR, Paludan SR. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J Immunol 2008; 181:8604-8612.
41. Zhou S, Halle A, Kurt-Jones EA, Cerny AM, Porpiglia E, Rogers M, Golenbock DT, Finberg RW. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2- MyD88/Mal-dependent inflammatory responses. J Neuroimmunol 2008; 194:70-82.
42. Gaudreault E, Fiola S, Olivier M, Gosselin J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol 2007; 81: 8016-8024.
43. Juckem LK, Boehme KW, Feire AL, Compton T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol 2008; 180:4965-4977.
44. Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol 2009; 83 (3):1492-1500.
45. Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 2007; 109: 619-625.
46. Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 2004; 127:1513-1524.
47. Aravalli RN, Hu S, Lokensgard JR. Inhibition of toll-like receptor signalling in primary murine microglia. J Neuroimmune Pharmacol 2008; 3:5-11.
48. Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol 2006; 177: 7094-7102.
49. Bauernfeind F, Hornung V. TLR2 joins the interferon gang. Nat Immunol 2009; 10:1139-1141.
50. Dolganiuc A, Chang S, Kodys K, Mandrekar P, Bakis G, Cormier M, Szabo G. Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol 2006; 177:6758-6768.
51. Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol 2007; 82:479-487.
52. Moriyama M, Kato N, Otsuka M, Shao R, Taniguchi H, Kawabe T, Omata M. Interferon-beta is activated by hepatitis C virus NS5 Band in habited by NS4A, NS4B, and NS5A. Hepatol. Int 2007; 1:302-310.
53. Riordan S, Skinner N, Kurtovic J, Locarnini S, McIver C, Williams R, Visvanathan K. Toll-like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm. Res 2006; 55:279-285.
54. Wang JP, Zhang Y, Wei X, Li J, Nan XP, Yu HT, Li Y, Wang PZ, Bai XF. Circulating toll-like receptor (TLR) 2, TLR4, and regulatory T cells in patients with chronic hepatitis C. APMIS 2010; 118:261-270.
55. Chung H, Watanabe T, Kudo M, Chiba T. Correlation between hyporesponsiveness to toll-like receptor ligands and liver dysfunction in patients with chronic hepatitis C virus infection. J. Viral Hepat 2011; 18(10): 561-567.
56. Brown R, Gralewski J, Eid A, Knoll B, Finberg R, Razonable R. R753Q single-nucleotide polymorphism impairs toll-like receptor 2 recognition of hepatitis C virus core and non structural 3 proteins. Transplantation 2010; 89: 811-815.
57. Organización Mundial de la Salud (OMS). (Fecha de consulta: 19/03/2011). Disponible en: http://whqlibdoc.who.int/publications/2009/9789241547871_eng.pdf.
58. Centers for Disease Control and Prevention (CDC). Dengue Homepage. (Fecha de consulta: 12/07/2012). Disponible en: http://www.cdc.gov/dengue/epidemiology/index.html.
59. Durán A, Bermúdez J, Maldonado MB, Ochoa E, Alcocer S, Levy A, Márquez A, Bermúdez I, Gómez M, Gotera J, Valero N. Incidencia y circulación del virus dengue en el Estado Zulia, Venezuela (2009-2010). Ciencia 2012; 20(1):22-32.
60. Versteeg GA, Garcia-Sastre A. Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 2010; 13:508-516.
61. Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, Martínez-Sobrido L, Ashok M, Lipkin WI, García-Sastre A. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol 2005; 79:8004-8013.
62. Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J Virol 2010; 84:9760-9774.
63. Chang TH, Chen SR, Yu CY, Lin YS, Chen YS, Kubota T, Matsuoka M, Lin YL. Dengue Virus Serotype 2 Blocks Extracellular Signal-Regulated Kinase and Nuclear Factor-kB Activation to Down regulate Cytokine Production. PLoS ONE 2012; 7(8): e41635. (doi:10.1371/journal.pone. 0041635). Available from: URL: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0041635.
64. Matsumoto M, Seya T. TLR3: interferon induction by double stranded RNA including poly (I:C). Adv Drug Deliv Rev 2008; 60:805-812.
65. Prehaud C, Megret F, Lafage M, Lafon M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol 2005; 79:12893-12904.
66. Farina C, Krumbholz M, Giese T, Hartmann G, Aloisi F, Meinl E. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol 2005; 159:12-19.
67. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol 2002; 61:1013-1021.
68. Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol 2010; 20(1):4-22.
69. Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections? Virology 2004; 322:231-238.
70. Gowen BB, Hoopes JD, Wong MH, Jung KH, Isakson KC, Alexopoulou L, Flavell RA, Sidwell RW. TLR3 deletion limits mortality and disease severity due to Phlebovirus infection. J Immunol 2006; 177:6301-6307.
71. Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Núñez G, Curtis JL, Luker GD. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol 2008; 180:483-491.
72. Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2006; 2:e53. [10. 1371/journal.ppat.0020053]. Available from: URL: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1475659.
73. Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 2008; 8:911-922.
74. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 2004; 10:1366-1373.
75. Daffis S, Samuel MA, Suthar MS, Gale MJ, Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol 2008; 82:10349-10358.
76. Wilson JR, de Sessions PF, Leon MA, Scholle F. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J Viro 2008; 82:8262-8271.
77. Arjona A, Ledizet M, Anthony K, Bonafé N, Modis Y, Town T, Fikrig E. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J Immunol 2007; 179:8403-8409.
78. Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, von Bernuth H, Ku CL, Casrouge A, Zhang XX, Barreiro L, Leonard J, Hamilton C, Lebon P, Héron B, Vallée L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova JL. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007; 317:1522-1527.
79. Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K, Takada K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signalling from Toll-like receptor 3. J Exp Med 2009; 206:2091-2099.
80. Liang Z, Wu S, Li Y, He L, Wu M, Jiang L, Feng L, Zhang P, Huang X. Activation of Toll-Like Receptor 3 Impairs the Dengue Virus Serotype 2 Replication through Induction of IFN-b in Cultured Hepatoma Cells. PLoS ONE 2011; 6(8):e23346. (doi:10.1371/journal.pone.0023346). Available from: URL: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0023346.
81. Wong JP, Christopher ME, Viswanathan S, Karpoff N, Dai X, Das D, Sun LQ, Wang M, Salazar AM. Activation of toll-like receptor signaling pathway for protection against influenza virus infection. Vaccine 2009; 27:3481-3483.
82. Zhou H, Zhao J, Perlman S. Autocrine interferon priming in macrophages but not dendritic cells results in enhanced cytokine and chemokine production after coronavirus infection. MBio 2010; 1(4). pii: e00219-10. [doi: 10.1128/mBio.00219 -10]. Available from: URL: http://www. ncbi.nlm.nih.gov/pubmed/20978536.
83. Mazaleuskaya L, Veltrop R, Ikpeze N, Martin-Garcia J, Navas-Martin S. Protective role of Toll-like receptor 3-induced Type I interferon in murine coronavirus infection of macrophages. Viruses 2012, 4:901-923.
84. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol 2000; 1:398-401.
85. Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol 2001; 75:10730-10737.
86. Hutchens MA, Luker KE, Sonstein J, Nunez G, Curtis JL, Luker GD. Protective effect of Toll-like receptor 4 in pulmonary vaccinia infection. PLoS Pathog 2008; 4: e1000153. [doi:10.1371/journal.ppat. 100 0153]. Available from: URL: http://www. plospathogens.org/article/info%3Adoi%2F 10.1371%2Fjournal.ppat.1000153.
87. Schabbauer G, Luyendyk J, Crozat K, Jiang Z, Mackman N, Bahram S, Georgel P. TLR4/CD14-mediated PI3K activation is an essential component of interferon-dependent VSV resistance in macrophages. Mol Immunol 2008; 45:2790-2796.
88. Okumura A, Pitha PM, Yoshimura A, Harty RN. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J Virol 2010; 84(1): 27-33.
89. Suh HS, Zhao ML, Choi N, Belbin TJ, Brosnan CF, Lee SC. TLR3 and TLR4 are innate antiviral immune receptors in human microglia: role of IRF3 in modulating antiviral and inflammatory response in the CNS. Virology 2009; 392:246-259.
90. Pine SO, McElrath MJ, Bochud PY. Polymorphisms in toll-like receptor 4 and toll-like receptor 9 influence viral load in a seroincident cohort of HIV-1-infected individuals. AIDS 2009; 23:2387-2395.
91. Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N. Association between common Toll-like receptor 4mutations and severe respiratory syncytial virus disease. J Infect Dis 2004; 189:2057-2063.
92. Zhang Y, Lian JQ, Huang CX, Wang JP, Wei X, Nan XP, Yu HT, Jiang LL, Wang XQ, Zhuang Y, Li XH, Li Y, Wang PZ, Robek MD, Bai XF. Over expression of Toll-like receptor 2/4 on monocytes modulates the activities of CD4(+)CD25(+) regulatory T cells in chronic hepatitis B virus infection. Virology 2010; 397(1):34-42.
93. Chen YC, Wang SY, King CC. Bacterial lipopolysaccharide inhibits dengue virus infection of primary human monocytes/ macrophages by blockade of virus entry via a CD14-dependent mechanism. J Virol 1999; 73:2650-2657.
94. Cabrera-Hernández A, Thepparit C, Suksanpaisan L, Smith DR. Dengue virus entry into liver (HepG2) cells is independent of hsp90 and hsp70. J Med Virol 2007; 79:386-392.
95. Modhiran N, Kalayanarooj S, Ubol S. Subversion of innate defenses by the interplay between DENV and pre-existing enhancing antibodies: TLRs signalin Collapse. PLoS Negl Trop Dis 4(12): e924. [doi:10.1371/ journal.pntd.0000924]. Available from: URL: http://www.plosntds.org/article/info %3Adoi%2F10.1371%2Fjournal.pntd.0000924.
96. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004; 303:1526-1529.
97. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA 2004; 101 (15):5598-5603.
98. Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol 2002; 3(6):499. [doi:10.1038/ni0602-499]. Available from: URL: http://www.nature. com/ni/journal/v3/n6/ full/ni0602-499.
99. Gorden KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS. Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol 2006; 177:6584-6587.
100. Diebold SS. Recognition of viral single-stranded RNA by Toll-like receptors. Adv Drug Deliv Rev 2008; 60:813-823.
101. Town T, Bai F, Wang T, Kaplan AT, Qian F, Montgomery RR, Anderson JF, Flavell RA, Fikrig E. Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 2009; 30:242-253.
102. Finberg RW, Wang JP. Antiviral responses: different roles for different tolls. Immunity 2009; 30:173-175.
103. Welte T, Reagan K, Fang H, Machain-Williams C, Zheng X, Mendell N, Chang GJ, Wu P, Blair CD, Wang T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J Gen Virol 2009; 90 (11):2660-2668.
104. Meier A, Chang JJ, Chan ES, Pollard RB, Sidhu HK, Kulkarni S, Wen TF, Lindsay RJ, Orellana L, Mildvan D, Bazner S, Streeck H, Alter G, Lifson JD, Carrington M, Bosch RJ, Robbins GK, Altfeld M. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat Med 2009; 15(8):955-959.
105. Oh DY, Baumann K, Hamouda O, Eckert JK, Neumann K, Kücherer C, Bartmeyer B, Poggensee G, Oh N, Pruss A, Jessen H, Schumann RR. A frequent functional toll like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS 2009; 23:297-307.
106. Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev 2009; 61:195-204.
107. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004; 21(1): 107-119.
108. Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci USA 2004; 101:11416-11421.
109. Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol 2007; 81: 3170-3180.
110. Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O’Keeffe M, Hochrein H. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J Clin Invest 2008; 118:1776-1784.
111. Jiang W, Lederman MM, Mohner RJ, Rodriguez B, Nedrich TM, Harding CV, Sieg SF. Impaired naive and memory B-cell responsiveness to TLR9 stimulation in human immunodeficiency virus infection. J Virol 2008; 82:7837-7845.
112. Malaspina A, Moir S, DiPoto AC, Ho J, Wang W, Roby G, O’Shea MA, Fauci AS. CpG oligonucleotides enhance proliferative and effector responses of B Cells in HIV infected individuals. J Immunol 2008; 181:1199-1206.
113. Martinelli E, Cicala C, Van Ryk D, Goode DJ, Macleod K, Arthos J, Fauci AS. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci USA 2007; 104:3396-3401.
114. Bochud PY, Hersberger M, Taffé P, Bochud M, Stein CM, Rodrigues SD, Calandra T, Francioli P, Telenti A, Speck RF, Aderem A. Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS 2007; 21(4):441-446.
115. Xie Q, Shen HC, Jia NN, Wang H, Lin LY, An BY, Gui HL, Guo SM, Cai W, Yu H, Guo Q, Bao S. Patients with chronic hepatitis B infection display deficiency of plasmacytoid dendritic cells with reduced expression of TLR9. Microbes Infect 2009; 11(4):515-523.
116. Huang XX, McCaughan GW, Shackel NA, Gorrell MD. Up-regulation of proproliferative genes and the ligand/receptor pair placental growth factor and vascular endothelial growth factor receptor 1 in hepatitis C cirrhosis. Liver Int 2007; 27:960-968.
117. Schlender J, Hornung V, Finke S, Günthner-Biller M, Marozin S, Brzózka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol 2005; 79: 5507-5515.
118. Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med 2008; 205:1277-1283.
119. Equils O, Schito ML, Karahashi H, Madak Z, Yarali A, Michelsen KS, Sher A, Arditi M. Toll-like receptor 2 (TLR2) and TLR9 signalling results in HIV-long terminal repeat transactivation and HIV replication in HIV-1 transgenic mouse spleen cells: implications of simultaneous activation of TLRs on HIV replication. J Immunol 2003; 170(10):5159-5164.
120. Wang JP, Bowen GN, Padden C, Cerny A, Finberg RW, Newburger PE, Kurt-Jones EA. Toll-like receptor-mediated activation of neutrophils by influenza A virus. Blood 2008; 112(5):2028-2034.
