











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCION
El cáncer constituye un problema de salud mundial debido a un aumento de la población, al envejecimiento, la adopción de un estilo de vida en el que existen factores de riesgo como el hábito de fumar, el sedentarismo, una alimentación deficiente, la obesidad, etc. Tradicionalmente, se consideraba el cáncer como un proceso exclusivo del genotipo celular como único causante. Actualmente, se abarca como un sinergismo entre el genotipo celular, y el microambiente tumoral. Estudiar este proceso complejo como un todo es difícil, mientras que hacerlo por etapas más sencillas y estudiarlas por separado, permite obtener resultados parciales, que en conjunto facilitan el estudio del proceso. Actualmente, la tendencia se centra en adquirir la capacidad de proporcionar un tratamiento adecuado a cada paciente, lo que se denomina una medicina de precisión, asumiendo que la biología que tiene cada tumor es única para cada caso. Esto permitirá desarrollar fármacos dirigidos a blancos específicos en las células tumorales, evitando dañar las células no tumorales, y disminuyendo sus efectos secundarios 1. La progresión tumoral es un proceso complejo en el que están implicadas múltiples alteraciones genéticas que conducen a las células normales a malignizarse, a través de un proceso de transformación progresivo 2. Se han descrito ocho alteraciones fisiológicas características de las células tumorales, comunes en la mayoría de los tumores, que son: 1) autosuficiencia de las señales de crecimiento, 2) insensibilidad a las señales inhibidoras de crecimiento, 3) evasión de la muerte celular programada o apoptosis, 4) potencial replicativo ilimitado, 5) capacidad de angiogénesis, 6) capacidad invasiva y metastática, 7) reprogramación del metabolismo energético, y 8) evasión del sistema inmune 3.
Células epiteliales: El tejido epitelial recubre las superficies externas e internas del organismo y está formado por una o varias capas de células unidas entre sí. Presenta una estructura característica formada por capas de células epiteliales unidas entre sí mediante contactos celulares, entre los que se incluyen: uniones estrechas, uniones adherentes, uniones tipo comunicantes y desmosomas. Así mismo, las células se contactan con la membrana basal, a través de enlaces célula-sustrato, que las separa de las células del tejido conectivo denominado estroma. La polaridad ápico-basal es característica de las células epiteliales, ya que presentan su superficie apical hacia el lumen, y su superficie basal hacia la membrana basal. Esta polarización conlleva una localización asimétrica del núcleo celular sobre la superficie basal, y una mayor superficie apical donde están presentes proteínas de superficie celular, mediante las cuales se establecen los diferentes tipos de uniones con células epiteliales adyacentes 4.
Los carcinomas. Son los más comunes de todos los cánceres, y tienen su origen en células epiteliales. Presentan una gran complejidad estructural, donde se observan diferentes tipos celulares, incluyendo células normales o no neoplásicas, que constituyen el estroma y que representan alrededor del 90% de la masa tumoral, y las células tumorales o transformadas. Los tipos celulares que forman el estroma son fibroblastos, células endoteliales, células de músculo liso, adipocitos, macrófagos y linfocitos, entre otras. La comunicación entre los diferentes tipos celulares que forman el estroma y el epitelio se define como heterotípica, que es fundamental para mantener la arquitectura y estructura del tejido. Las células estromales y epiteliales colaboran en la formación de la matríz extracelular (MEC) 5.
Invasión y metástasis en carcinomas. Los carcinomas comienzan en la zona epitelial en contacto estrecho con la membrana basal, y se consideran benignos mientras que las células que lo forman no atraviesen la membrana basal. En algunos casos, los carcinomas adquieren la capacidad de romper la membrana basal, invadir el estroma y metastatizar; es a partir de este punto que se denominan malignos, y se considera a este tumor original, como un tumor primario. Para que ocurra la invasión local del estroma, se requiere la secreción de proteasas que degradan la MEC para generar espacios a través de los cuales pueden migrar las células tumorales. En algunos tipos de cánceres, las propias células tumorales secretan sus propias proteasas, mientras que, en otros tipos de tumores, son las células estromales las que secretan dichas proteasas 6.
Para que un tumor primario crezca, necesita que se desarrolle una red de vasos linfáticos y sanguíneos para cubrir las necesidades metabólicas de las células, como nutrientes y oxígeno. Esta nueva formación de vasos se denomina linfangiogénesis y angiogénesis, respectivamente, y la entrada de las células a los vasos linfáticos y sanguíneos ocurre mediante un proceso denominado intravasación, accediendo así al torrente circulatorio y permitiendo la diseminación de las células tumorales 7. En algunas ocasiones, estas células tumorales presentes en el sistema circulatorio, pueden penetrar en un tejido distal al tumor primario mediante un proceso denominado extravasación, y dar lugar a micrometástasis. Las células que forman parte de la micrometástasis pueden crecer, dividirse y formar una macrometástasis para alcanzar el estadio de colonización, y dar lugar a lo que se denomina tumor secundario 8. La probabilidad de que una célula tumoral complete todos los pasos descritos, desde la ruptura de la membrana basal, invasión hasta la formación de metástasis, es muy baja. Los primeros pasos son ejecutados con elevada eficiencia por las células metastáticas, no así los pasos finales que implican la colonización. Aún así, el hecho de que células neoplásicas circulen por el torrente circulatorio corporal se considera un factor de mal pronóstico para el paciente. Las metástasis son responsables de alrededor del 90% de las muertes por cáncer, por eso es importante un diagnóstico precoz, así como el estudio de nuevas terapias que bloqueen los estadios tempranos del proceso de invasión y metástasis 9.
Transición epitelio-mesénquima: fisiológica y patológica
La progresión tumoral de carcinomas se caracteriza por la capacidad de invasión por parte de las células tumorales epiteliales que sufren múltiples alteraciones al someterse a un proceso denominado “Transición epitelio-mesénquima (TEM)”, y que tiene lugar en estadios tempranos de la invasión y la metástasis. Durante la TEM en cáncer, las células epiteliales pierden su fenotipo epitelial y polarizado 10,11. Este proceso está acompañado de una reorganización del citoesqueleto celular y de una pérdida de adhesión célula-célula y célula-sustrato, que dan lugar a una morfología mesenquimal, que proporciona a las células una capacidad invasiva y de metástasis, pudiendo migrar por el torrente sanguíneo a órganos distales diferentes al tumor primario 12,13.
La TEM fue inicialmente descrita por Elisabeth Hay, durante el desarrollo embrionario, como un paso clave para la formación de órganos y tejidos 14. La TEM se ha observado en los procesos de regeneración tisular, fibrosis, y cáncer. El proceso de TEM se ha descrito en tres marcos biológicos muy diferentes, donde la generación de células mesenquimales es común para todos ellos, mientras que el proceso biológico a través del cual se adquiere el fenotipo mesenquimal es muy diferente. Se conocen tres tipos de TEM propuestos en función del proceso biológico: Tipo I: asociada con la implantación, la formación embrionaria y el desarrollo de órganos. Todos estos procesos precisan generar diferentes tipos celulares, que comparten fenotipos mesenquimales comunes. Tipo II: asociada con la cicatrización de heridas, regeneración tisular, inflamación y fibrosis, este proceso genera fibroblastos y otros tipos celulares relacionados para regenerar el tejido dañado después de un traumatismo o una inflamación. Tipo III: asociada a la conversión de células epiteliales en células mesenquimales, que pueden migrar e invadir tejidos, favoreciendo la progresión tumoral 11.
Transición epitelio-mesénquima (TEM) y cáncer
La adquisición de capacidades propias de células mesenquimales por parte de las células epiteliales, implica una progresión maligna de las mismas a través de un proceso biológico en el que se suprimen los marcadores celulares epiteliales, y donde las células pierden su polaridad, y las uniones célula-célula. Esto provoca cambios en la morfología celular y en el citoesqueleto, lo que hace que las células pierdan el fenotipo epitelial y adquieran una morfología similar a fibroblastos, adquiriendo capacidades de migración e invasión celular 15. Estos cambios están acompañados por la pérdida de marcadores epiteliales en las células, tales como caderina-E, zonula occludens-1 (ZO- 1), citoqueratinas (proteína fibrosa de los filamentos intermedios del citoesqueleto), así como por un incremento de los marcadores mesenquimales que incluyen caderina-N, vimentina, actina de músculo liso, y proteína-1 específica de fibroblastos, además de la producción de componentes de matríz extracelular como colágeno tipo I y fibronectina (glucoproteina) 16 (ver Fig. 1). La desestabilización del complejo caderina-E/catenina, provoca una acumulación citoplasmática de catenina, que es capaz de translocarse al núcleo donde actúa como activador de Tcf-1/ LEF (factores de transcripción involucrados en la vía de señalización Wnt), y por consiguiente comienzan a expresarse genes que codifican proteínas con un importante papel oncogénico, tales como ciclina D1(proteínas que controlan la progresión de la fase G1 a la fase S) y c-Myc (protooncogen), originando la inducción del proceso TEM 17. No está claramente definido el total de señales que contribuye al proceso de TEM asociado al cáncer. Una hipótesis plantea que mediante las comunicaciones heterotípicas originadas por las células del estroma asociados al tumor, se inducen las alteraciones genéticas y epigeneticas, que sufren las células tumorales durante el proceso de formación del tumor primario. Además, las células estromales también pueden secretar factores de crecimiento, tales como el factor de crecimiento hepático (HGF) 18, el factor de crecimiento epidérmico (EGF) 19, el factor de crecimiento derivado de plaquetas (PDGF) 20, y el factor de crecimiento transformante beta (TGβ) 21, que pueden inducir la expresión de los factores de transcripción Snail, Slug, ZEB-1, ZEB-2 y Twist 22,23. Estos factores de transcripción actúan como represores transcripcionales de la caderina-E, originando por consecuencia, la pérdida de uniones entre las células epiteliales y de la polaridad apical. Durante la progresión tumoral la perdida de la caderina-E va acompañada de la expresión de las caderinas mesenquimales caderina-N, caderina-P, caderina-6, caderina-11 (glucoproteína transmembrana) 24. En cáncer de próstata y de mama la sobre-expresión de caderina-N promueve la motilidad y la migración celular 25. En el cáncer colorrectal se ha observado un significativo incremento de la expresión de caderina-N comparado al tejido adyacente sano, y se ha relacionado con una baja diferenciación tumoral, y una baja supervivencia 26. El intercambio de caderina-E por caderina-N en cáncer epitelial de vejiga se ha propuesto como un factor de malignidad que identifica a las células tumorales epiteliales de vejiga con un fenotipo invasivo 27. Así mismo, la caderina-P y la caderina-11 se expresan en cáncer de mama invasivo, mientras que la cadherina-6 se encuentra sobreexpresada en carcinoma renal 28.
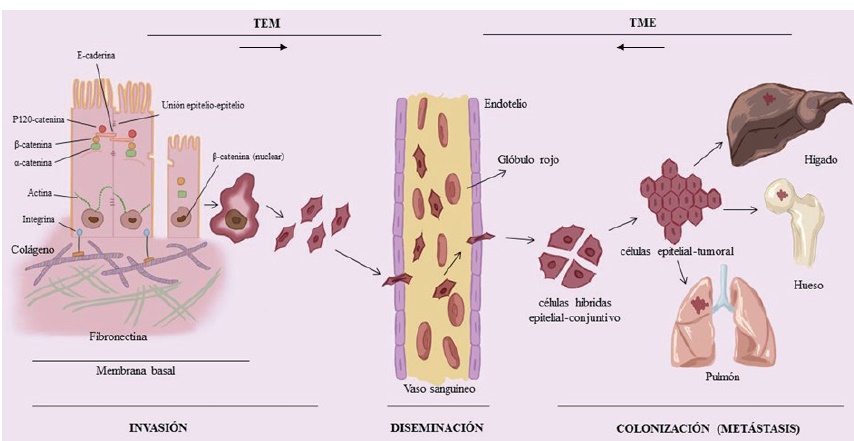
Plasticidad epitelial: implicaciones clínicas
La TEM es un proceso transitorio y reversible, es decir, las células epiteliales que han adquirido capacidades y fenotipos mesenquimales pueden revertir el proceso, y volver a su estado epitelial original diferenciado, a través del procedimiento denominado Transición mesénquima-epitelio (TME), que es transitorio y reversible, y que recibe el nombre de plasticidad epitelial 29,30 (Fig. 1). Clínicamente, el proceso de TEM tiene lugar en las células del carcinoma primario, probablemente en estadios tempranos de la invasión y la metástasis. La TEM en la progresión tumoral está en discusión, porque la identificación y detección de células tumorales circulantes (CTCs), que sufren un proceso de TEM en el torrente sanguíneo de pacientes oncológicos es muy difícil, debido a que se encuentran presentes en muy bajo número. Las CTCs son células tumorales que se desprenden del tumor primario y viajan por el torrente sanguíneo hasta alcanzar otras partes del cuerpo originando metástasis tumorales 31. Las metástasis tumorales son las causantes de la mayoría de las muertes por cáncer. El proceso biológico de la metástasis es muy poco eficaz, ya que menos del 0.01% de las células presentes en el tumor primario es capaz de acceder al torrente sanguíneo y dar lugar a las metástasis 32. Se considera que las metástasis clínicas ocurren incluso antes de detectar el tumor primario, lo que sugiere que el proceso de TEM ha tenido lugar antes de que el tumor crezca en el órgano primario 33. Terapéuticamente, hay diferentes estrategias para poder impedir la formación de metástasis a través de la acción sobre la TEM: a saber 1) Inhibir la inducción de la TEM, 2) Promover la TME, 3) eliminar o inhibir funcionalmente a las células tumorales mesenquimales 34. En cáncer de ovario, una de las primeras estrategias para bloquear la metástasis fue inhibir el proceso de TEM para prevenir la diseminación de células tumorales desde el tumor primario 35. Se ha observado que la inducción de la TEM conduce a la colonización, crecimiento y formación de tumores secundarios y por consiguiente a la micrometástasis 36; por lo que en principio, el objetivo fue bloquear aquellas células que se diseminaban y así evitar su desplazamiento y el inicio de la metástasis. Al iniciarse el bloqueo muchas de estas células, ya estarían trasladándose a los tejidos blanco, producto de su liberación del tumor primario en las etapas tempranas del desarrollo tumoral. Por tal motivo, el tumor primario no sería el objetivo sino el nicho metastásico, evitando el alojamiento, división y formación de nuevos tumores al bloquear las señales de crecimiento que evitaría la colonización a otros tejidos 13.
Uniones adherentes: caderinas-E
La caderina-E, es la mejor caracterizada de las uniones adherentes en células epiteliales de mamíferos. La caderina-E es un supresor de tumores, y su pérdida de expresión es un marcador de TEM, y está asociada a estadios tempranos de invasión y metástasis, a un menor grado de diferenciación, y a un mal pronóstico clínico 37. La pérdida de regulación de la caderina-E en las uniones adherentes es importante, ya que marca los estadios tempranos de cáncer antes que ocurra la metástasis. Es importante destacar que las células epiteliales deben mantenerse unidas entre sí para estructurar el epitelio que formará parte de un tejido polarizado e íntegro en su estructura 37.
Estructura de la caderina-E
La caderina-E es el prototipo mejor caracterizado de la familia de las células epiteliales. Se expresa en todos los epitelios presentes en mamíferos, y funcionalmente, se ha relacionado con el mantenimiento de la polaridad ápico-basal y la preservación de la supervivencia, y del control de la proliferación celular. Esta proteína está codificada por el gen cadherin 1 (CDH1) presente en el brazo largo del cromosoma 16. El dominio citoplasmático se divide en 1) dominio de juxtamembrana, y 2) dominio de unión a catenina 38. Se ha propuesto que la regulación del mantenimiento de caderina-E en las uniones célula-célula, se realiza a través del bloqueo de la interacción de miosina II, proteína motora dependiente de actina, con caderina-E evitando de esta manera su disgregación 39.
Inhibidores de microtúbulos y transición epitelio-mesénquima
El citoesqueleto celular está formado por el citoesqueleto de actina, los filamentos intermedios y la red de microtúbulos, y juega un papel fundamental en el proceso
de TEM. La reorganización del citoesqueleto de actina durante la progresión tumoral, es el paso clave en la adquisición de capacidades migratorias e invasivas de las células tumorales que promueven la metástasis celular. Se ha observado una sustitución de la citoqueratina presente en los filamentos intermedios por vimentina, durante la TEM 40. Los microtúbulos proporcionan la fuerza motora necesaria para la migración celular durante la progresión tumoral, los cuales se distribuyen uniformemente en el citoplasma de las células epiteliales no transformadas, mientras que las protusiones de la membrana celular, debido a TEM son estructuras asociadas a microtúbulos 41. Dada la función de los microtúbulos en el crecimiento y la proliferación celular, son considerados blancos para el empleo de compuestos que inhiben la polimerización de los microtúbulos en el tratamiento de pacientes con cáncer 42. Los compuestos antimitóticos que se fijan a microtúbulos se clasifican en dos grupos: 1) agentes estabilizadores de microtúbulos, y 2) agentes desestabilizadores de microtúbulos que actúan inhibiendo la polimerización, como la vinflunina y el nocodazole. Por otra parte, el compuesto quinolina-6-il oxyacetamidas, identificado inicialmente como un fungicida frente a un amplio rango de fitopatógenos, tamnién actúa como un desestabilizador de microtúbulos, con propiedades antiproliferativas en células tumorales de pulmón y ovario resistentes al tratamiento con otras drogas antineoplásicas 43,44. Las drogas que inhiben la actividad de los microtubulos puede tener un efecto potencial sobre la adhesión celular 45.
Regulación de la caderina-E
En la mayoría de los cánceres humanos de origen epitelial, se ha demostrado a menudo la pérdida de caderina-E 46. Los niveles de caderina-E para mantener la adhesión celular dependen del balance entre la síntesis y la degradacion 47. Existe una correlacion inversa entre los niveles de caderina-E, el estadio del tumor y la tasa de mortalidad, aunque en algunos tumores diferenciados se expresa la caderina-E 48. La restitución de los complejos formados por la interacción de caderina-E con catetina-β, catetina-∑ (proteína intracitoplasmatica), y p120, induce a una reversión del fenotipo mesenquimal hacia el fenotipo epitelial de células tumorales en cultivo celular 49.
Los reguladores de caderina-E son:
Reguladores genéticos o epigenéticos. En tumores de mama y ovario se han identificado alteraciones genéticas del gen CDH1, que codifica para caderina-E, que causan la pérdida de su funcionalidad 50. La región del promotor de CDH1 que corresponde a las islas CG, sufre una metilación anormal o hipermetilación, ocasionando pérdida de la expresión de caderina-E 51.
Reguladores transcripcionales. La acción de diferentes represores transcripcionales de caderina-E durante la TEM, como Snail, Slug, Twist, ZEB-1, y ZEB-2, da origen a la pérdida de expresión de la caderina-E 52.
Reguladores post-traduccionales. La regulación post-traduccional se realiza a través de la fosforilación, la glicosilación o la proteolisis. La fosforilación de los resíduos de tirosina de la caderina-E, está mediada por el EGF y el factor de crecimiento de fibroblastos (FGF), responsable de la fosforilación de caderina-E, y relacionado con la pérdida de caderina-E 53. La proteína caseína quinasa 1 (CK1) es un regulador negativo de la caderina-E que participa en la fosforilación de resíduos en su dominio citoplasmático 54. Diferentes metaloproteasas de la matríz (MMP), como MPP2, MPP9 y MPP14, participan en la degradación de la caderina-E. La caderina-E está representada como una molécula soluble de 80 kDa, que se encuentra en células tumorales en cultivo y en las biopsias tumorales 55. En el cáncer nasofaríngeo la sobreexpresión de MMP-2 inhibe la adhesión célula-célula, promoviendo la TEM e invasión tumoral, con una baja regulación de caderina-E y alta regulación de caderina-N, fibronectina y Slug 56.
Migración de las células mesenquimáticas
Una vez que ha ocurrido la TEM, las células mesenquimáticas invaden la membrana basal y sintetizan matríz de fibronectina, que proporciona una vía para la migración de estas células mesenquimales durante la TEM, manteniendo el fenotipo mesenquimal 57. La presencia de fibronectina en la MEC aumenta la rígidez del sustrato, 58 controlado por las señales SMAD (factor de transcripción) y JNK (quinasa) activadas por TGF-β (factor de crecimiento β) 59. Una mayor cantidad de fibronectina junto con Ras (reguladoras de los procesos de transducción) activado en las células epiteliales, provoca el reemplazo de las integrinas a6b3 por las a5b1 que mejoran la capacidad migratoria de las células, aumentando su adhesión celular a la fibronectina 60. El aumento de colágeno tipo I y fibronectina en la MEC, también se correlaciona con el cambio de las integrinas a un estado de unión a ligandos de alta afinidad, aumentando la actividad de señalización a través de FAK (proteína de anclaje) e ILK (integrina), promoviendo la TEM 61. La diversidad en las afinidades de unión de diferentes integrinas, permite a las células responder a una amplia gama de elementos extracelulares, y mediar en diferentes cascadas de señalización en respuesta a un entorno de matríz cambiante. A medida que cambia la composición de la MEC, también lo hace la abundancia de integrinas en la superficie celular, lo que promueve la progresión de la TEM bajo el control del entorno pericelular. Las integrinas aVb3 facilitan la fosforilación de TGF-βRII mediada por Src (proto-oncogen), originando un sitio de acoplamiento para ShcA (receptor nuclear) y GRB2 (proteína de receptor de crecimiento), que envían señales a través de la vía p38 MAPK (proteínas quinasa) para inducir TEM 62. La importancia del colágeno tipo I en la TEM es evidente en varios sistemas celulares. El colágeno tipo I se asocia con la TEM en los carcinomas de pulmón, mama y páncreas, lo que destaca la importancia del entorno de la matríz en la metástasis 63. La inducción de TEM depende de la interacción entre las fibras de colágeno tipo I y la integrina a2b1, lo que desencadena una cascada intracelular 64. El colágeno de tipo I provoca la fosforilación de IkB (inhibidor de kB) dependiente de ILK para aumentar la abundancia de NF-κB (factor de transcripción) de localización nuclear, que promueve la expresión de SNAI1 y LEF1 (proteína en células B y T) para inducir TEM 65. El aumento de colágeno tipo I activa las vías de JNK, donde se ha demostrado que la inhibición farmacológica de la señalización de JNK anula la migración mediada por colágeno tipo I, y la metástasis de las células en cáncer de mama 66,67.
Heterogeneidad y resistencia
La heterogeneidad biológica que se manifiesta a través de mecanismos tanto genéticos como no genéticos, contribuye a las diferencias fenotípicas entre las diferentes subpoblaciones de células cancerosas que residen en tumores individuales. Se reconoce la capacidad de las células cancerosas para adquirir estados fenotípicos alternativos, sin cambios mutacionales en su genoma. El concepto de células madres cancerosas (CMC) postula la presencia de poblaciones menores de CMC que tienen la capacidad de sembrar nuevos tumores. Existen mecanismos reguladores epigenéticos que pueden contribuir a la diversidad fenotípica de distintas subpoblaciones de células cancerosas dentro de un tumor. Esto se basa en el concepto de que células cancerosas fenotípicamente distintas que residen dentro de la misma masa tumoral están organizadas en jerarquías, semejante a la jerarquía de células madres del tejido no neoplásico. En los tumores las células con fenotipo CMC deberían en principio, renovarse por sí mismas, para generar nuevas CMC que se diferencien en una descendencia menos tumorigénica y no autorrenovadora; es decir, las no CMC que forman la mayor parte del tumor. El concepto de CMC tal vez define la eficacia a menudo limitada de las terapias convencionales contra el cáncer, debido a que se centran en la población mayor de células no CMC dentro de los tumores individuales y no eliminando específicamente las CMC. Las evidencias experimentales han demostrado que las CMC son más resistentes que las no CMC. Esto demuestra la capacidad de las CMC para ser precursoras de nuevas masas tumorales, lo que trae como consecuencia la recaída clínica. En los cánceres epiteliales, estos cambios adaptativos pueden involucrar, al menos en parte TEM y el proceso inverso TME. La TEM puede desencadenar la reversión a un fenotipo similar a CMC, 68,69 lo que proporciona una asociación entre TEM, CMC y resistencia a los fármacos que origina cambios fenotípicos sin que ocurran cambios genéticos. Se ha observado una relación directa entre TEM y resistencia a quimioterapia, además de la importancia del proceso de TEM en la invasión y la metástasis. Existen múltiples estudios clínicos que ensayan compuestos farmacológicos cuyo blanco terapeútico son genes implicados en el proceso de plasticidad epitelial. La detección de altos niveles de Zeb-1 y Twist, represores transcripcionales de caderina-E, y por consiguiente reducción de caderina-E y otros marcadores epiteliales que se relaciona con la resistencia al tratamiento con diferentes drogas, como son 5-fluorouracilo, gemcitabina o cisplatino 70. La disminución en la expresión de caderina-E se utiliza en el ámbito clínico como factor del mal pronóstico, ya que se relaciona con una baja diferenciación epitelial de las células tumorales, y se asocia a procesos de invasión y metástasis que implican baja supervivencia en diferentes tipos de cáncer tales como el cáncer de mama o cáncer colorrectal 71. En el cáncer colorrectal (CCR) una de las neoplasias malignas más agresivas, la LACTB (serina proteasa mitocondrial que actúa como regulador del metabolismo de los lípidos mitocondriales) funciona como un supresor de tumores. Se ha demostrado que LACTB puede inhibir la TEM y la proliferación del cáncer de mama. LACTB (serina proteasa) regula el nivel de PIK3R3 (fosfatidil-inositol 3-quinasa) para promover la autofagia e inhibir la TEM y la proliferación. Estos efectos se logran en parte a través de la vía de señalización PI3K /AKT (oncogen de retrovirus murino)/ mTOR (proteína quinasa) en el CCR 72.
Mecanismo de resistencia a fármacos inducida por TEM
Durante mucho tiempo se ha tratado de establecer una relación entre TEM y la resistencia a los medicamentos, pero hasta el presente no se ha dilucidado un mecanismo concreto. Probables evidencias se han obtenido de los trabajos con las CMC, con los cuales se tienen perspectivas sobre el mecanismo de resistencia a los fármacos en células sometidas a TEM. Las CMC son una subpoblación de células que forma parte de la masa tumoral responsable de la tumorigénesis 73. Se han encontrado similitudes en las vías de señalización de Wnt, Hedgehog y Notch (vías de señalización que regulan la proliferación, diferenciación y migración) en el fenotipo activado TEM y CMC. Estas vías son críticas para el mantenimiento y renovación de CMC 74. Esto sugiere que las células sometidas a TEM presentan propiedades parecidas a las células madres al compartir vías de señalización de fenotipo resistente a fármacos 75,76. La terapia del cáncer está asociada con la quimioresistencia y la recurrencia después de la quimioterapia 77,78. Esta resistencia se asocia a su vez, con la presencia, dentro de los tumores, de poblaciones agresivas de células cancerosas que desarrollaron mecanismos de quimioresistencia que conducen a una disminución de las tasas de supervivencia de pacientes tratados por cáncer 79. En varios estudios, se han identificado estas poblaciones de cánceres agresivos como CMC 80,81. La quimioresistencia mediada por CMC se basa en varios mecanismos celulares:
1. Baja tasa de proliferación. La quimioterapia se dirige a las células altamente proliferativas, a través del daño en el ADN e inhibición de la división mitótica; su acción es limitada cuando se aplica a células cancerosas lentas y que no se dividen, como las CMC 82,83. Algunos estudios han demostrado que las CMC de diferente origen tisular, podrían resistir el efecto de una amplia gama de fármacos como la doxorrubicina, temozolomida, cisplatino, paclitaxel, etopósido y metotrexato 84. Por lo tanto, el desarrollo de terapias que se dirijan a las células inactivas o de división lenta como CMC es esencial para prevenir cánceres recurrentes.
2. Expresión de transportadores de “casette” de unión a ATP (ABC). Esta gran superfamilia de proteínas de membrana, funciona en el transporte transmembrana de varios sustratos mediante la conversión de la energía obtenida de la hidrólisis del ATP85 Aunque, el tipo de transportadores ABC varía de un tipo de cáncer a otro, las CMC expresan altos niveles de estos transportadores, lo que contribuye a un mayor transporte de compuestos quimioterapéuticos fuera de las células, contribuyendo así a la quimiorresistencia de las CMC. Por ejemplo, el transportador ABCC1 conocido como MRP1 (proteína de resistencia a múltiples fármacos 1), expresado por CMC en glioblastoma, está involucrado en la salida de una variedad de compuestos terapéuticos que incluyen: metotrexato, edatrexato, ZD1694, doxorrubicina, daunorrubicina, epirrubicina, idarrubicina, etopósido, vincristina, vinblastina, paclitaxel, irinotecán, SN-38, flutamida e hidroxiflutamida 86. Por otra parte, el transportador MDR1 (proteína de resistencia a múltiples drogas 1), expresado igualmente por CMC’ se encuentra en cánceres de ovario y de mama, leucemia mieloide aguda (LMA), glioblastoma y carcinoma de células renales, proporciona multirresistencia a fármacos como las antraciclinas, actinomicina D, colchicina, etopósido, tenipósido, metotrexato, mitomicina C, mitoxantrona, paclitaxel, docetaxel, vincristina y vinblastina 87. Hay otros transportadores ABC con capacidad como ABCA1 (transportador de casette de unión al ATP A1), que se expresa en las CMC de ovario y que transporta cisplatino 88. En las células sometidas a TEM con sobreexpresión de los transportadores ABC y mostraron un fenotipo de resistencia a fármacos similares a las CMC. Saxena y col. en 2011, demostraron que los promotores de los transportadores ABC, contienen varios sitios de unión para TEM-TF. La sobreexpresión de TEM-TF como Twist, Snail y FOXC2 (factor de transcripción), aumentó la actividad promotora y la expresión de transportadores ABC en células de cáncer de mama. Estas células mostraron una resistencia diez veces mayor al tratamiento con doxorrubicina comparadas con el control 89.
3. Aumento de la expresión de aldehído deshidrogenasas (ALDH). Esta super-familia de enzimas es determinante para la desintoxicación de sustratos de aldehídos endógenos y exógenos al catalizar la oxidación de aldehídos a ácidos carboxílicos 90. Las ALDH están altamente expresadas en CMC de diferentes tipos de cánceres, y sus niveles elevados de expresión se correlacionan con un deficiente pronóstico en pacientes con cáncer 91. Además, están asociadas con la quimiorresistencia mediada por CMC, aunque sus mecanismos de acción no han sido bien dilucidados 92.
4. Resistencia a la apoptosis. Las CMC resisten la apoptosis mediada tanto por la vía de muerte intrínseca (dependiente de mitocondrias), como por la vía de los receptores de muerte celular extrínseca 93. Por ejemplo, los CMC de glioma y leucemia humanos, expresan bajos niveles de Fas y ligando de Fas (Fas-L) (proteínas de membranas), lo que da como resultado resistencia a la muerte celular mediada por receptores extrínsecos 94. Además, de la vía apoptótica extrínseca, la proteína pro-supervivencia Bcl-2 está involucrada en la vía de muerte dependiente de mitocondrias, a través de sus interacciones inhibidoras con los proapoptóticos Bax y Bak. La proteína Bcl-2 (proto-oncogen), se encontró sobreexpresada en células madre de leucemia, glioma y glioblastoma 95. La inactivación de las vías de muerte celular intrínsecas y extrínsecas, asegura una ventaja de supervivencia selectiva para las CMC.
5. Respuesta de reparación del ADN. Otra ventaja de supervivencia está relacionada con la capacidad de las CMC para activar oportunamente el sensor de daño del ADN 96. Este proceso involucra vías de reparación del ADN que incluyen la reparación por escisión de nucleótidos (REN), reparación por escisión de bases (REB), reparación de errores de apareamiento (MMR), reparación directa y reparación de rotura de doble hebra (DSB). La evidencia de la resistencia de CMC a la radioterapia, fue proporcionada por un estudio que demostró la capacidad de las células madre de glioblastoma para activar eficientemente el ATM de la serina/treonina quinasa y el daño en el ADN de la proteína quinasa de control (Chk1) en respuesta a las radiaciones ionizantes 97.
6. Microambiente CMC. Los nichos de células madre representan áreas de tejido que proporcionan microambientes específicos, que mantienen y promueven la capacidad de las CMC para autorrenovarse y generar progenies diferenciadas 98. El nicho de células madre es necesario para determinar el destino de estas células, ya que su comportamiento está influenciado por su asociación con otras células del nicho. Este concepto es aplicable a las CMC, donde la interacción con estos nichos es necesaria para el mantenimiento de las poblaciones de CMC. El microambiente es un complejo altamente heterogéneo compuesto por células tales como células estromales, células inmunes y células epiteliales, y una red de macromoléculas extracelulares que proporciona soporte a las células dentro de la MEC 99. Las células que se encuentran en el nicho promueven el crecimiento, el mantenimiento y la diferenciación de las CMC. Las terapias contra el cáncer no han tenido éxito, ya que los fármacos eliminan la población masiva de células cancerosas, sin afectar a las poblaciones de CMC 100.
7. Fibroblastos asociados al cáncer (FAC). Expresan un apoyo mecánico para las CMC a través de la producción de colágeno fibrilar. Los FAC fibroblastos asociados al cancer (FAC), también secretan la citocina CXCL12 (ligando 12 de quimiocina del motivo CXC), factores de crecimiento como el HGF, el factor de crecimiento endotelial vascular (VEGF) y el factor de crecimiento derivado de plaquetas (PDGF), que contribuyen al aumento de la proliferación, invasión y metástasis de las CMC 101. Los FAC participan en la heterogeneidad celular a través del TGFβ1, que promueve la TEM relacionada con CMC 102.
8. Células inmunes. Las células inmunes contribuyen al estado inflamatorio crónico del microambiente de las CMC, que potencia la proliferación, invasión y metástasis tumorales 103. Los macrófagos asociados a tumores (TAM) y las células supresoras derivadas de mieloides (MDSC) secretan TGF-β, que contribuye a la TEM, la invasión y las metástasis 104. Las células inmunes del microambiente contribuyen a la evasión tumoral a través de una variedad de mecanismos. Los TAM, a través del TGFβ reclutan células T reguladoras (Tregs) dentro del nicho y contribuyen a la inmunosupresión. Las MDSC, secretan factores de crecimiento como TGFβ, y las citocinas reclutan células T auxiliares para promover su actividad inmunosupresora 105.
9. Células madre mesenquimales (CMM). Son células estromales multipotentes que pueden diferenciarse en osteocitos, adipocitos y condrocitos. Estas células migran a sitios inflamatorios crónicos como es el caso del cáncer, donde contribuyen a la metástasis al secretar TGFβ que promueve la TEM 106. También participan en la colonización de cáncer secundario en cánceres metastásicos de mama, próstata y pulmón 107. Las CMM promueven la proliferación del cáncer gástrico y la angiogénesis a través de la secreción de VEGF, proteína 2 inflamatoria de macrófagos (MIP-2), TGF-β1 y las citocinas proinflamatorias interleucinas IL -6 e IL -8 108. Las CMM pueden originar FAC, que contribuyen aún más a la heterogeneidad de las células del microambiente CMC y a su potencial metastásico 109,110.
10. Células endoteliales. La angiogénesis es clave para el microambiente de CMC, ya que proporciona nutrientes para el metabolismo, que son necesarios para su autorrenovación y capacidades invasivas y metastásicas. A través de los vasos tumorales, las células inmunitarias como las Treg, contribuyen a la supresión inmunitaria 111. Las células endoteliales, junto con las células perivasculares, constituyen los componentes básicos de los vasos, y son estimuladas por factores angiogénicos como el VEGF dentro de un entorno hipóxico como el cáncer. Esto resulta en un aumento de la vasculatura del tumor y un mayor crecimiento y metástasis 112. Además, las células endoteliales tumorales secretan citocinas como IL -3, granulocitos (globulos blancos)-CSF (factores estimuladores de colonias de células sanguíneas), granulocitos-macrófagos-CSF (GM-CSF), IL -1, IL -6, VEGF-A y bFGF (factor de crecimiento de fibroblastos), que promueven y conservan la autorrenovación y la progresión mediada por CMC 113.
11. Hipoxia. En el cáncer, la hipoxia ocurre como resultado de condiciones isquémicas 114. Por lo tanto, la baja tensión de oxígeno es capaz de provocar cambios en el fenotipo celular y cooperar con otras vías para inducir la TEM. La TEM inducida por hipoxia, está estrechamente mediada por la vía de señalización de HIF (factor de transcripción), que contribuye al crecimiento tumoral agresivo y a la invasividad. Estudios experimentales han demostrado que el crecimiento en condiciones hipóxicas, puede reprogramar las células epiteliales a un fenotipo mesenquimatoso, debido a la activación de represores de la transcripción de cadherina-E, lo que conduce a la promoción del potencial invasivo 115. Se han propuesto varios aspectos de los posibles mecanismos moleculares. En primer lugar, la activación de HIF-1 y 2 puede inducir TEM mediante la regulación positiva de factores de transcripción asociados a TEM o represores como Twist, Snail, Slug y SIP1/ZEB2 (factor de transcripción) en células cancerosas 116,117. En segundo lugar, la hipoxia y la vía HIF activan las vías de señalización asociadas a TEM como TGF-β, Notch, NF-κB, Wnt/catetina-β y Hedgehog 118,119. La hipoxia y la vía de HIF pueden inducir el fenotipo o las características de la TEM, mediante la regulación de las citocinas inflamatorias asociadas a la TEM, como el aumento de la expresión del factor de necrosis tumoral α inducido por hipoxia (TNF-α), la interleucina 6 (IL -6) y IL -1β, que promueve la inducción del fenotipo TEM 120,121. Así mismo, la hipoxia y la vía de HIF pueden inducir la TEM mediante la regulación directa o indirecta de proteínas o enzimas que median las interacciones célula-matriz, y facilitan la motilidad y la invasión mediadas a través de la regulación de LOX / LOX2 (lisil oxidasa), Hey1, Hes1 (represores transcripcionales) y el activador del plasminógeno tipo uroquinasa (uPA) 122,123.
12. Estroma. El estroma desempeña un papel en la protección de las CMC contra la quimioterapia y otros tratamientos, proporcionando a las CMC, estímulos de señalización resistentes a través de receptores de superficie para activar otras líneas de defensa. Los miofibroblastos tumorales, secretan factores de crecimiento como el HGF y la periostina, para activar la señalización de Wnt en el cáncer colorrectal y el de mama 124,125. Las células estromales secretan altos niveles de HGF, lo que hace que las células cancerosas humanas de muchos tipos co-cultivadas, adquieran resistencia a varios fármacos, en particular a los de RAF (proteína quinasa) 126. Otros factores de crecimiento o citocinas, incluidos la IL -6, el FGF y la neuregulina 1, ayudan a formar el llamado “nicho quimiorresistente” de las CMC, activando diversas vías de señalización de supervivencia 127. Además, las integrinas de los receptores de MEC, están altamente asociadas con la supervivencia de la CMC y la resistencia a fármacos, lo que representa un papel crítico para el MEC 128. El marcador de superficie de células madre CD44, inicialmente identificado como el receptor de localización de linfocitos, se expresa comúnmente en células madre embrionarias y adultas, así como en las CMC. Esto ocurre porque se une a su ligando hialuronano para regular muchos aspectos de la función de las células madre, incluida su autorrenovación. La señalización de CD44 también ayuda a mantener la integridad genómica de las células madre al protegerlas y reparar el ADN después del daño oxidativo. Además, CD44 está implicado en la regeneración de CMC responsables de la quimiorresistencia 129.
13. Canales iónicos que promueven TEM. El microambiente hipóxico además de promover programas de supervivencia adaptativos en el cáncer, se asocia con la resistencia, la apoptosis, la angiogénesis y la migración, e induce la TEM en una variedad de tipos de células cancerosas 130. El calcio (Ca2+) es un mensajero intracelular que desempeña un papel crucial en el cáncer, incluída la proliferación 131. La angiogénesis y la metástasis, también están involucradas en la inducción de TEM a través del EGF como una consecuencia de la entrada de Ca2+ a la célula 132. El microambiente del tumor es rico en ATP, por lo tanto es probable que aumenten los niveles de Ca2+ libre intracelular, mediante la activación de los receptores purinérgicos 133 o indirectamente, a través de la activación de proteínas G y la generación de trifosfato de 1, 4, 5 inositol (IP3) por el receptor P2Y 134.
El canal iónico del potencial receptor transitorio canónico-1 (TRPC1), es un componente clave de las respuestas a la hipoxia en las células de cáncer de mama. Esta regulación incluye el control de eventos específicos de TEM, y la activación de vías de señalización mediadas por hipoxia, como la activación del EGFR (receptor del factor de crecimiento epidermal), STAT3 (citoquina) y el marcador de autofagia LC3B, a través del factor inducible por hipoxia-1α (HIF1α). El receptor transitorio canonico-1 (TRPC1), regula los niveles de HIF1α en las líneas celulares de cáncer de mama MDA-MB-468 y HCC1569 deficientes en PTEN. Esta regulación surge de los efectos sobre la traducción constitutiva de HIF1α en condiciones normóxicas a través de una vía dependiente de Akt. En apoyo adicional al papel de TRPC1 en TEM, su expresión está estrechamente asociada con genes relacionados con TEM y metástasis en tumores de mama. La expresión de TRPC1 también es de pronóstico significativo para los cánceres de mama basales, en particular los clasificados como positivos para los ganglios linfáticos. Los roles definidos de TRPC1 se podrían investigar terapéuticamente para el control de rutas oncogénicas en células de cáncer de mama 135.
14. Hormonas
Insulina. La señalización del factor de crecimiento similar a la insulina (IGF), está constituida por una red dinámica de proteínas que incluye los ligandos (IGF-I e IGF-II) y sus receptores asociados, proteínas de unión a IGF (IGFBP) y proteasas de IGFBP 136. La proteína IGF-1, ha estado más fuertemente implicada en la progresión del cáncer de mama debido a su efecto mitogénico y antiapoptótico sobre las células epiteliales mamarias 137. El IGF-I puede actuar de forma endocrina, paracrina o autocrina. Por consiguiente, los niveles y la actividad de IGF-1 se han examinado de cerca en tejidos proliferativos, para estudiar su relación con los cambios en la morfología celular asociados con la progresión del cáncer. La insulina puede estimular la síntesis del factor de crecimiento similar a la insulina 1 (IGF-1), y ambos tienen potentes efectos mitogénicos sobre las células tumorales. La insulina y el IGF-1 pueden activar las vías PI3K/Akt/ mTOR y Ras/Raf/MAPK, estimulando así el crecimiento tumoral 138. Estos productos génicos dan como resultado la activación extracelular de MMP. Las MMP junto a la MEC pueden activar a TGF-β1. El TGF-β1 aumenta la producción de la matríz extracelular, como la fibronectina, como parte de su papel en la inducción de TEM. Por otra parte, la IGFBP-3 inducida por TGFβ puede actuar como un promotor tumoral en presencia de fibronectina, ya que podría mediar en la actividad pro-tumorigénica de TGFβ para inducir TEM 139. En ratones inmunosuprimidos, las células epiteliales mamarias humanas sobreexpresaron IGF-IR, que se asoció con el inicio de TEM, mediante la regulación positiva de Snail y la regulación negativa de caderina-E 140. En base a la sobreexpresión del IGF-IR, se asocia con un fenotipo agresivo en una variedad de tumores 141.
Leptina. Es pro-tumorigénica, actúa sobre las células epiteliales de mama promoviendo la proliferación a través de la señalización de estrógenos, la activación de MAPK y STAT3, y la inhibición de la apoptosis mediante la activación de AKT 142. Además, es pro-angiogénica e induce la TEM en el cáncer de mama, además promueve la auto-renovación tanto de las CMC como del cáncer de mama 142. La leptina participa en la señalización para la activación de un circuito proinflamatorio que promueve la progresión del cáncer. Se ha demostrado que la interferencia entre IL -1, leptina y Notch promueve la proliferación, la migración y la regulación positiva del factor de crecimiento endotelial vascular y del receptor 2 en el cáncer de mama 143. Wang y col. en 2015, señalaron que la leptina promueve la TEM en las células del cáncer de mama a través de la regulación positiva de IL -8, mediante la activación de la vía de señalización PI3K/Akt 144. La leptina también promueve la migración e invasión de las células del epitelio mamario mediante la activación de un complejo regulador formado por las cinasas FAK y Src, que favorecen la expresión de las proteínas relacionadas con la formación de estructuras proteolíticas, implicadas en la invasión y progresión del cáncer. Tizapa y col. en 2019, señalaron que la sobreexpresión y activación de la proteína Hic-5 durante la TEM, favorece la formación de invadopodios, promoviendo la degradación de los componentes de la MEC e induciendo la metástasis del cáncer 145.
Adiponectina. Tiene un papel importante en el metabolismo de la glucosa, los lípidos, la sensibilidad a la insulina, la angiogénesis, la inmunidad, la inflamación y el cáncer 146. La adiponectina inhibe la proliferación de células de cáncer de mama con receptores estrógenos negativos, a través de la señalización de MAPK y AKT, promoviendo la apoptosis y regulando negativamente las propiedades metastásicas, al inhibir la señalización de mTOR mediante la activación del complejo enzimático (AMPK), mientras que a bajas concentraciones podría promover el crecimiento y la progresión del tumor en cáncer de mama con receptores de estrógenos positivos 147,148. Funciona también como potencial supresor de tumores, inhibiendo la TEM, pero es frecuentemente silenciada en el cáncer de próstata por hipermetilación del promotor 149. Existen estudios que han demostrado la asociación inversa entre la adiponectina y el riesgo de cáncer de próstata o cáncer de próstata de grado alto 149,150. Así mismo, desempeña un papel en el bloqueo de la carcinogénesis, mediante la inhibición de la proliferación y la promoción de la apoptosis, e inhibe el VEGFα, evitando así, la neovascularización del cáncer 151. La adiponectina provoca la detención del ciclo celular de las líneas de células del estroma y del epitelio prostático e induce la apoptosis aumentando la caspasa-3 y regulando negativamente el gen Bcl2 (linfoma de células B) 152. En el cáncer de pulmón, la adiponectina puede ejercer un efecto antiproliferativo a través de la regulación por disminución de CREB (proteína involucrada en la tumorigénesis y significa: proteína de unión al elemento de respuesta a AMPc). Se demostró que las concentraciones fisiológicas disminuían significativamente la proliferación celular del adenocarcinoma de pulmón humano 153. En un estudio de cáncer de tiroides, Dossus y col. en 2018, detectaron bajos niveles de adiponectina circulante, que generalmente acompaña a la obesidad, y que se asocian con un mayor riesgo de cáncer de tiroides 154. Porcile C y col. en 2014, demostraron que la adiponectina inhibía la proliferación celular de las líneas celulares de glioblastomas humanos, al inducir la detención del crecimiento dentro de la fase G1. Además, observaron que regulaba negativamente la acción del factor de crecimiento similar a IGF-1, aboliendo la proliferación inducida por IGF-1 de las líneas celulares de glioblastoma 155.
Estrógeno. El estrógeno E2 (estradiol) induce la TEM. Durante la progresión del tumor, el estrógeno puede fomentar la motilidad celular y la invasión del cáncer de mama ER positivo al promover la TEM 156. La señalización ERα puede regular la caderina-E y TEM a través de Slug 157. El ERβ (receptor de estrógeno β) inhibe TEM en el cáncer de próstata desestabilizando el factor de transcripción inducible por hipoxia e inhibiendo la localización nuclear de Snail, mediada por el factor de crecimiento endotelial vascular 158. El Midkine (MK), factor de crecimiento que se une a la heparina, es capaz de ejercer actividades como la proliferación celular, la migración celular y la angiogénesis, también puede inducir TEM a través de la señalización Notch2/Jak2-Stat3(citoquinas) en queratinocitos humanos 159 e impulsar la TEM en las células de cáncer de páncreas, mediante la activación de la señalización Notch 160. EL MK también se induce fuertemente durante la oncogénesis, la inflamación y la reparación de tejidos inducida por E2. En las líneas celulares de adenocarcinoma de pulmón LTEP-a2 and A549, E2, regula el incremento de la expresión de MK. Así mismo, E2 indujo el reclutamiento de ERβ como elemento de la respuesta a estrógenos (ERE) en el promotor MK. La transcripción de MK inducida por E2 fue mediada principalmente por ERβ, lo que sugiere que la MK inducida por E2 juega un papel importante en la progresión de la TEM. Resultados similares de los niveles de proteínas relacionados con E2, MK y TEM fueron obtenidos en tejidos clínicos del adenocarcinoma de pulmón 161.
Testosterona. En el cáncer de vejiga, la expresión del receptor de andrógenos (RA), es más elevada, además la expresión de RA aumenta con la etapa del tumor, especialmente en el tejido del cáncer metastásico, lo que señala que RA representa un factor crítico en el desarrollo del cáncer de vejiga 162. Se demostró que TEM desempeña un papel vital en la metástasis del carcinoma urotelial, relacionado a través de VEGF, EGF y TGF-β 163. El TGF-β puede regular RA para activar la TEM, proporcionando evidencias para que RA pueda considerarse como una terapia contra el cáncer urotelial. La acción de los andrógenos se ejerce a través del eje que implica la síntesis testicular de testosterona, su transporte a los tejidos diana y su conversión por la 5-reductasa en el metabolito activo 5-dihidrotestosterona (DHT). Los andrógenos ejercen sus efectos biológicos al unirse al RA e inducir su actividad transcripcional. Peng y col. en 2006, demostraron que la inactivación de la expresión de RA en las células T24 por micro-ARN inhibió la capacidad de migración de estas células. Esto puede estar asociado con una disminución de las concentraciones de MMP-9, que resultó del silenciamiento del gen RA. Las MMP promueven la progresión del cáncer y la metástasis al impulsar el crecimiento, la migración, la invasión y la angiogénesis 164. Por otra parte, los andrógenos estimulan la expresión de MMP-9 en células de cáncer de próstata dependientes de andrógenos y la eliminación de RA suprimió las propiedades invasivas de estas células, debido a la disminución de la expresión de MMP-9 165. Jitao y col. en 2014, utilizando la línea celular de vejiga T24, determinaron que el silenciamiento de RA por micro-ARN podría suprimir la progresión del cáncer de vejiga de manera significativa tanto in vitro como in vivo. Realizaron estudios de silenciamiento para regular la expresión del RA y determinar la participación de RA en TEM, en presencia o ausencia de un RA-micro-RNA. Los resultados demostraron que RA indujo el patrón TEM en las células tumorales de vejiga y condujo a cambios significativos en la migración de células cancerosas de vejiga y en el potencial de invasión. La supresión de los niveles de expresión de RA, se correlacionó con la TEM mediada por andrógenos en las células T24, y en las cuales TGF-β puede desempeñar un papel importante, por lo que RA podría tener un gran potencial en la terapia del cáncer de vejiga 166. Se ha identificado al RA como un represor de caderina-E y podría activar la TEM en células tumorales y la metástasis en el cáncer de mama 167,168. Por otra parte, los andrógenos podrían inducir las características de TEM y la reorganización del citoesqueleto a través de la interacción con la señalización de TGF-β, lo cual podría contribuir al comportamiento metastásico del cáncer de próstata resistente a la castración 169. Además, se demostró que existía una regulación recíproca entre RA y Slug en células de cáncer de próstata, lo que indica un papel importante de la señalización RA en TEM, y que Slug es un gen regulado por andrógenos en las células de cáncer de próstata 170. En base a la señalización de RA en cáncer de vejiga, y el cual representa un factor crítico para TEM y metástasis, Jing y col. en 2014, en estudios tanto “in vitro” como “in vivo”, demostraron que los andrógenos regulan positivamente la expresión de Slug e inducen la TEM en células de cáncer de vejiga RA-positivas, a través de la activación de la vía Wnt/β-catenina. Así mismo, además de Slug, se determinó la expresión de Snail, Twist y ZEB-1 con el tratamiento de DHT en células cancerosas de vejiga RA-positivas. Los resultados mostraron una regulación positiva significativa para Slug, pero no de los otros factores de transcripción. Esto es consistente con el hallazgo de que la expresión de RA en los tejidos del cáncer de vejiga, muestra una alta correlación con los niveles de expresión de Slug 171. Slug es un miembro de la familia Snail de factores de transcripción de dedos de zinc, que ha sido identificado como un potencial oncogén en varios tipos de tumores, y que es capaz de reprimir la expresión de caderina-E y activar la TEM 172.
CONCLUSIÓN
Es fundamental que los inductores de TEM se mantengan en silencio para mantener la homeostasis y la integridad del epitelio. La aparición de TEM puede considerarse como la reactivación de programas del desarrollo similares que operan a nivel celular, aunque en este caso en el cáncer, genera consecuencias fatales. Es importante precisar cuáles son las señales micro ambientales inductoras de TEM, que permiten cambios en las células, que las hacen sensibles a tales señales, y determinar los mecanismos de señalización dentro de las células epiteliales que ordenan a los diversos programas de TEM. Obtenido este conocimiento, puede ser trasladado a la práctica clínica a través de la oncología de precisión que se basa en analizar el perfil genético, clínico y molecular del paciente, para posteriormente poder definir un tratamiento personalizado, con las mayores posibilidades de éxito, contribuyendo de esta forma a mejorar su calidad de vida y su supervivencia.

