










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.24 n.1 Caracas 2005
Resistencia a drogas en M. Tuberculosis: Bases moleculares
N Arráiz1, V Bermúdez2, B Urdaneta3.
1 Doctor en Ciencias Biológicas. Centro de Investigaciones Endocrino-Metabólicas "Dr. Félix Gómez". Sección de Biología Molecular La Universidad del Zulia. Facultad de Medicina, Maracaibo-Venezuela
2 Doctor en Ciencias Médicas. Centro de Investigaciones Endocrino-Metabólicas "Dr. Félix Gómez". Sección de Biología Molecular La Universidad del Zulia. Facultad de Medicina, Maracaibo-Venezuela
3 Especialista en Ginecología y Obstetricia. Centro de Investigaciones Endocrino-Metabólicas "Dr. Félix Gómez". Sección de Biología Molecular La Universidad del Zulia. Facultad de Medicina, Maracaibo-Venezuela
RESUMEN
La tuberculosis es una de las enfermedades infecciosas responsables de altas tasas de morbi-mortalidad a nivel mundial. La terapia de corta duración (DOTS) recomendada por la Organización de la Salud (OMS) incluye los antibióticos isoniazida, rifampicina, pirazinamida, estreptomicina y etambutol. Lamentablemente, la estrategia DOTS deja de ser la opción terapéutica para pacientes infectados con M. tuberculosis multirresistentes a drogas (TB-MDR) definidas como cepas resistentes al menos a isoniazida y rifampicina. La resistencia a drogas en M. tuberculosis se debe predominantemente a alteraciones en genes que codifican blancos de antibióticos y hasta el presente se han identificado múltiples mutaciones cromosomales asociadas al desarrollo de resistencia a antibióticos de primera línea. La caracterización de estas mutaciones ha conducido al desarrollo de nuevas estrategias de diagnóstico molecular que podrían acortar el periodo de reporte del patrón de resistencia a drogas en M. tuberculosis. La detección temprana de cepas resistentes a drogas de primera línea, contribuiría a un mejor manejo terapéutico del paciente con drogas de segunda línea y disminución del riesgo de propagación de cepas de TB-MDR.
Palabras Clave: DOTS, M. tuberculosis, TB-MDR, Rifampicina, Resistencia, Antibióticos.
ABSTRACT
The tuberculosis is one of the infectious diseases responsible for high rates of morbi-mortality at world-wide level. The therapy of short duration (DOTS) recommended by the Organization of the Health (WHO) includes drugs isoniazid, rifampin, pyrazinamide, streptomycin and ethambutol. Unfortunately, strategy DOTS stops being the therapeutic option for patients infected with multidrug-resistant M.tuberculosis (MDR-TB) defined as TB resistant to at least isoniazid and rifampicin. The resistance to drugs in M. tuberculosis predominantly derives from alterations in genes that encodes antibiotic targets and until the present multiple chromosomal mutations associated to the development of resistance to first line drugs have been identified. The characterization of these mutations has lead to the development of new strategies for molecular diagnosis that could shorten the period of report of the drugs resistance pattern in M. tuberculosis. The early detection of resistant to first line drugs, provides useful insights for better therapeutic management of patient with second line drugs and decreasing the risk of propagation of TB-MDR strains.
Key Words: DOTS, M. tuberculosis, MDR-TB, Rifampicin, Resistance, Antibiotics.
INTRODUCCIÓN
La tuberculosis ha reemergido como un problema de salud pública de gran magnitud. A mediados de la década pasada se reportaron a nivel mundial aproximadamente 8 millones de nuevos casos, con una mortalidad de 3 millones anuales(1-4) y se ha estimado que un tercio de la población mundial está infectada con M. tuberculosis(5). En el octavo informe anual de la Organización Mundial de la Salud (OMS) del año 2004, la cifra de nuevos casos de tuberculosis se incrementó 8,8 millones para el 2002, de los cuales 3,9 millones fueron bacilíferos (Tabla 1)(6). La OMS ha alertado que si no se toman medidas urgentes de control, para el periodo comprendido entre el año 2002 y 2020, se podrían alcanzar cifras de 1000 millones de nuevos casos de personas infectadas con M. tuberculosis, de los cuales 150 millones desarrollarían la enfermedad y 36 millones morirían por esta causa(6).

En los años 50 la quimioterapia redujo exitosamente la incidencia y la transmisión de tuberculosis, sin embargo a partir de la década de los 80, se declara una emergencia a nivel mundial debido a la aparición de cepas resistentes a drogas antimicrobianas(5,7). A partir de 1990 surge lo que ha sido denominado por algunos autores "la nueva tuberculosis", alertando la emergencia y propagación de cepas de micobacterias resistentes a múltiples antibióticos, particularmente a drogas antituberculosas de primera línea, problemática que se ha visto agravada por el incremento de infecciones micobacterianas en pacientes coinfectados con el virus de la inmunodeficiencia humana (VIH), lo cual ha complicado la epidemiología de la tuberculosis(5-7).
Desde 1991, la OMS estableció la terapia multidroga debido a la aparición de cepas mutantes de M. tuberculosis con resistencia a algunos de los antibióticos utilizados para el momento, bajo la hipótesis de que la cepa resistente podía ser controlada, al incluir al menos otros dos componentes antimicrobianos(8-10).
Las cepas de M. tuberculosis que son resistentes a isoniazida y rifampicina, con o sin resistencia a otras drogas, han sido denominadas cepas multirresistentes a drogas o TB-MDR(7,10). La OMS estima que 50 millones de personas a nivel mundial podrían estar infectadas con TB-MDR y que aproximadamente 3,1% de los 8,8 millones de nuevos casos en el año 2002 fueron causados por TB-MDR(6-7,10). Los regímenes terapéuticos de pacientes infectados con cepas TB-MDR son menos efectivos, más prolongados, de difícil cumplimiento y más costosos(11-15).
El resurgimiento de la tuberculosis con características epidémicas y la aparición de cepas multirresistentes, hace imperativo el diseño de estrategias de diagnóstico y estudios de susceptibilidad a drogas que permita obtener resultados inmediatos(12,14,16). Sin duda, el conocimiento de las bases moleculares de los mecanismos de resistencia a antibióticos provee información de gran valor para el manejo del paciente en varios aspectos(17). El análisis genético de los microorganismos podría acortar el periodo de reporte de resistencia a drogas, particularmente a drogas de primera línea, con la finalidad de adoptar un esquema terapéutico acertado y oportuno y así, lograr la cura del paciente, lo que se traduce en una disminución en la transmisión del patógeno y de la propagación de cepas multirresistentes a drogas. Adicionalmente la caracterización molecular del mecanismo de resistencia puede brindar información sobre el potencial de virulencia de una cepa particular y por último el análisis de alteraciones moleculares representa la base para el diseño de nuevas drogas(17,18).
TRATAMIENTO DE TUBERCULOSIS
El tratamiento de tuberculosis se basa en un régimen terapéutico cuádruple con antibióticos de primera línea y la mayoría de los países siguen los lineamientos de la Organización Mundial de la Salud a través de la estrategia DOTS (directly observed treatment, short course)(6,8,9). Los antibióticos de primera línea tienen propiedad bactericida e incluyen: isoniazida (H), rifampicina (R), pirazinamida (Z) y estreptomicina (S). Para el tratamiento de casos nuevos se incluye etambutol (E), el cual aunque es bacteriostático, tiene la propiedad de prevenir la resistencia a fármacos de primera línea.
La terapia estándar de corta duración o DOTS recomendada por la OMS para tratar pacientes con cepas sensibles, consiste en una fase inicial de dos meses con los antibióticos isoniazida, rifampicina pirazinamida y etambutol o estreptomicina, seguida de una fase de tratamiento continuo de cuatro meses con isoniazida y rifampicina. Este esquema terapéutico de seis meses de duración alcanza tasas de curación superiores al 90% y regularmente se reportan bajos niveles de resistencia(6,11-13). Según el último informe de la OMS, de un total de 210 países, 180 (85%) aplican la estrategia DOTS a través de los Programas Nacionales de Tuberculosis (PNT) y un 70% de la población mundial vive en estos países(6).
En pacientes con fracasos en el tratamiento, el régimen de la OMS consiste en la administración de isoniazida, rifampicina y etambutol por un periodo de 8 meses, junto con pirazinamida durante los primeros tres meses y estreptomicina los primeros dos meses(9,15). Lamentablemente, los cultivos y las pruebas de susceptibilidad a antibióticos no siempre están disponibles y se corre el riesgo de administrar este tratamiento a pacientes infectados con TB-MDR, en cuyo caso estaríamos en presencia de una monoterapia con etambutol(19).
Cuando se sospecha de TB-MDR, no puede obviarse el cultivo y las pruebas de susceptibilidad a drogas, sin embargo se comienza a administrar al paciente un tratamiento empírico basado en drogas de segunda línea hasta que se obtengan los resultados de las pruebas de susceptibilidad (Tabla 2), enfatizando que no debe agregarse una única droga al protocolo terapéutico inicial y que se deben incluir al menos tres drogas no utilizadas previamente(6,15,20).
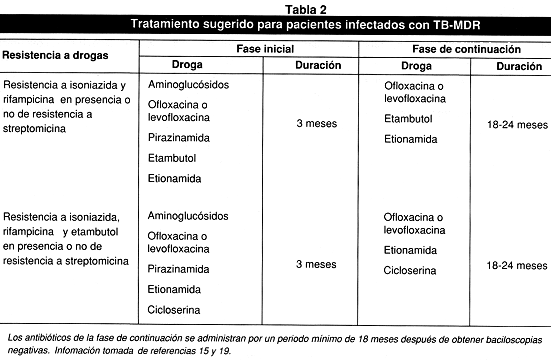
Los pacientes infectados con cepas resistentes a drogas, deben ser investigados en el laboratorio para ser tratados bajo el esquema DOTS-PLUS(12), con nuevos antibióticos o antibióticos de segunda línea para los cuales se haya demostrado sensibilidad en pruebas de susceptibilidad a antibióticos en el laboratorio y que no hayan sido administrados al paciente con anterioridad(6,12,13). Los principales fármacos de segunda línea que se utilizan para el tratamiento de pacientes con TB-MDR, son amikacina (A), protionamida/etionamida (Pt/Et), capreomicina (CM), kanamicina (K), cicloserina (CS), fluoroquinolonas, ácido r-aminosalicílico (PAS) y tioacetazona (Tabla 2). Estos exhiben menos efectividad y una gran variedad de efectos adversos, deben ser administrados con mayor frecuencia que los antibióticos de primera línea y el promedio de duración del tratamiento recomendado es de dos años (Tabla 2), haciendo más difícil el cumplimiento de la terapia por parte del paciente(10,13,20).
Una de las limitaciones de la mayoría de los programas de control de tuberculosis es que el diagnóstico se basa fundamentalmente en la baciloscopia con tinción Ziehl-Neelsen, por las restricciones de tiempo y costos que representan el aislamiento primario de la bacteria, dificultando así la detección de casos de resistencia a antibióticos. La resistencia a drogas se infiere cuando el paciente no responde al tratamiento después de dos meses de terapia, lo cual trae como consecuencia deterioro del paciente o muerte y mayor oportunidad de desarrollo de resistencia a otras drogas con el consecuente riesgo de propagación de cepas TB-MDR en la población(10). Como es de esperar, el esquema terapéutico DOTS fracasa en pacientes infectados con TB-MDR definidas como cepas resistentes al menos a rifampicina e isoniazida(10,11-13).
A través de diversos estudios en todo el mundo se ha encontrado que las cepas resistentes a rifampicina ya han desarrollado resistencia a otros antibióticos, por lo cual la característica de resistencia a rifampicina se considera un marcador para la detección de cepas de Mycobacterium tuberculosis MDR(17-19). La importancia de la detección de cepas TB-MDR, utilizando rifampicina como marcador reside en que la terapia de corta duración DOTS propuesta por la OMS deja de ser la mejor opción para el tratamiento de pacientes infectados con TB-MDR y en la medida que la detección de una cepa multirresistente se haga mas temprano, mayor será la probabilidad de indicar un esquema terapéutico adecuado y oportuno(15,16,18-20). Así, la caracterización y comprensión de los mecanismos moleculares de resistencia a drogas ha conducido al desarrollo de nuevas estrategias diagnósticas y terapéuticas.
DEFINICIÓN Y CAUSAS DE RESISTENCIA A DROGAS
Las cepas de M. tuberculosis resistentes a drogas se definen como tal por su capacidad para crecer a mayores concentraciones de antibióticos que las cepas sensibles. Las cepas silvestres son aquellas que nunca han estado en contacto con drogas antituberculosas, mientras que las cepas con resistencia natural son cepas silvestres resistentes a una droga dada, sin haber estado expuestas a la misma(10,11).
La resistencia primaria se refiere a infección de un paciente que nunca ha recibido tratamiento, por una cepa resistente. Ésta incluye infección por cepas silvestres que nunca han estado en contacto con drogas (resistencia natural) y también la resistencia que se desarrolla como consecuencia de la exposición de una cepa a determinada droga, pero en otro paciente. Se define como resistencia secundaria o adquirida, aquella que se desarrolla en pacientes que han recibido quimioterapia antituberculosa, debido a la selección de cepas mutantes resistentes espontáneas, en la mayoría de los casos, motivado a un tratamiento inadecuado o incumplimiento de la terapia(14,15). La resistencia primaria es informativa de deficiencias y fracasos en los programas de control de la tuberculosis, mientras que la resistencia adquirida es indicativa de deficiencias en el tratamiento actual de un paciente con tuberculosis(11,14-16).
Otros conceptos importantes en el manejo del paciente y la epidemiología de TB-MDR son la monorresistencia y polirresistencia. La OMS define la monorresistencia como resistencia de M. tuberculosis a uno de los cinco antibióticos de primera línea, mientras que polirresistencia se refiere a resistencia a dos o más antibióticos de primera línea(10,12,13). Multirresistencia a drogas o TB-MDR, como se definió anteriormente, es un tipo de polirresistencia en el cual M. tuberculosis desarrolla resistencia al menos a rifampicina e isoniazida, las drogas más efectivas en la terapia antimicobacteriana, en ausencia o presencia de resistencia a otras drogas(7,10,13).
El éxito de la estreptomicina en el tratamiento de la tuberculosis en 1943 pronto se vio opacado por la aparición de cepas resistentes al antibiótico. Actualmente, se conoce que la monoterapia conduce a la emergencia de cepas resistentes a una tasa de 1:106, al menos a una de las drogas conocidas(14), de manera que un paciente tratado con un solo antibiótico tiene una alta probabilidad de seleccionar mutantes resistentes(14-16). Por ejemplo, la tasa de mutación para desarrollar resistencia a isoniazida se ha estimado en 1:106 replicaciones y la tasa de resistencia a rifampicina en 1:108 replicaciones, de manera que existe una alta probabilidad de desarrollar resistencia a alguno de estos antibióticos en una lesión pulmonar que generalmente contiene 107-109 bacilos, sin embargo, la probabilidad de desarrollar resistencia a los dos antibióticos se reduce a 1:1014 (17-19).
Debido a que la tasa de mutación a dos o más drogas es más baja, se hizo evidente la necesidad de aplicar protocolos de tratamientos con múltiples drogas, por ejemplo introduciendo ácido r-aminosalicílico (1946) e isoniazida (1952) y consistente con esto, la terapia multidroga es exitosa e impide el desarrollo de resistencia en pacientes que son tratados por primera vez(7,8,11,14). Así, se introdujeron tratamientos combinados introduciendo otros antibióticos efectivos contra M. tuberculosis, tales como pirazinamida en 1952, cicloserina en 1955, etambutol en 1961 y rifampicina en 1966(7,11,12). El uso de la terapia combinada de rifampicina y pirazinamida permitió la disminución del periodo de tratamiento desde 18-24 meses a 6 meses(13,14,16).
Aunque se ha establecido el éxito de la terapia multidroga en el tratamiento de tuberculosis y en consecuencia, la disminución de la transmisión de cepas resistentes, también se ha advertido que siguen emergiendo cepas multirresistentes debido a monoterapias no intencionales que resultan de una inadecuada prescripción de antibióticos que no se ajustan a los protocolos recomendados, pobre adherencia del paciente a la terapia, fallas en la supervisión del tratamiento, limitado suministro de drogas, bajo estándar en la calidad de las drogas antituberculosas y en general, fallas en las políticas nacionales para apoyar los programas de control de tuberculosis(6).
DROGAS DE PRIMERA LÍNEA Y MECANISMOS MOLECULARES DE RESISTENCIA EN M. TUBERCULOSIS
La resistencia a drogas en M. tuberculosis se debe predominantemente a alteraciones en la secuencia de nucleótidos en genes que codifican blancos de antibióticos y a diferencia de otras bacterias, en M. tuberculosis no se han reportado mecanismos de adquisición de genes de resistencia vía plásmidos o transposones. Las micobacterias del complejo tuberculosis desarrollan resistencia a múltiples drogas por la acumulación de mutaciones individuales en varios genes, cada uno de los cuales es responsable de la resistencia a un antibiótico particular. A continuación se describen las alteraciones génicas asociadas al desarrollo de resistencia a antimicrobianos de primera línea (Tabla 3).

Resistencia a Isoniazida (INH)
La isoniazida (ácido nicotínico hidrazida o INH) tiene acción bactericida al interferir con la biosíntesis de ácidos micólicos(18-20). Es una prodroga que al ser captada por el bacilo, es activada por el sistema catalasa-peroxidasa, de manera que la ausencia de actividad catalasa, debido a mutaciones en el gen katG, codificante de esta enzima, es uno de los mecanismos de resistencia a INH(17,20-22). Las cepas de M. tuberculosis con mutaciones en el gen katG exhiben poca o ninguna actividad catalasa y son altamente resistentes a INH (MIC >32 mg/ml). Las mutaciones se concentran en una región codificante del gen katG, que comprende los codones 300 al 507, siendo las más frecuentes las sustituciones de la serina 315 por treonina (S315®T) y el residuo de arginina 463 por leucina (R463®L)(23,24). Estas mutaciones explican aproximadamente el 50% de los casos de aislados clínicos resistentes a INH (Tabla 3)(21,23).
Por otra parte, la enzima enoil ACP reductasa, involucrada en los pasos de elongación de ácidos grasos, codificada por gen inhA se identificó como un blanco de acción de INH(25,26). El intermediario de INH, cuya activación depende de la actividad catalasa-peroxidasa intacta, inhibe la actividad de la enzima inhA y en consecuencia la síntesis de ácidos micólicos(25). Las mutaciones en el gen inhA inducen sobreexpresión del gen inhA y niveles elevados de la enzima enoil reductasa en cantidades que superan el poder inhibitorio de INH. Las mutaciones en inhA están asociadas a aproximadamente al 25% de los casos de resistencia a INH, generalmente con bajos niveles de resistencia (MIC£1mg/ml)(26,27).
La investigación de otros genes involucrados en resistencia a INH que explicaran el mecanismo de resistencia del 10-20% de cepas que carecían mutaciones en katG o inhA condujo a la identificación del gen ahpC, codificante de la enzima alquil hidroperóxido reductasa(27-29), involucrada en la respuesta a estrés oxidativo. Las mutaciones en aphC están asociadas a aproximadamente un 10 a 15% de aislados clínicos resistentes a INH y actualmente se investigan otros genes candidatos asociados con resistencia a este antibiótico(29).
Resistencia a Rifampicina (RIF)
La rifampicina es un antibiótico de amplio espectro clave en la estrategia DOTS propuesta por la OMS y el mecanismo de resistencia a este antibiótico es uno de los primeros caracterizados a nivel molecular. El mecanismo de acción de la rifampicina consiste en que el antibiótico se une a la ARN polimerasa procariota, la enzima responsable del proceso de transcripción de genes, y al inhibir la expresión de genes, la rifampicina conduce a la muerte de la célula. La resistencia a rifampicina se explica por mutaciones en el gen rpoB, el cual codifica la subunidad b de la ARN polimerasa y las alteraciones en esta subunidad impiden que la rifampicina interactúe adecuadamente con la ARN polimerasa e inhiba la transcripción(24,27,30).
Se ha demostrado que la resistencia a rifampicina en M. tuberculosis se explica en un 95% a 98% por mutaciones en el gen rpoB, las cuales generalmente se localizan en un corto segmento de aproximadamente 81 pb que incluye los codones 507 a 533 del gen rpoB(27,30-32). Las mutaciones en esta región incluyen delecciones, inserciones, sustituciones, siendo las más frecuentes, las mutaciones en codones para asparagina 516, histidina 526 y serina 531(30-34), de manera que los métodos genotípicos para ensayar resistencia a rifampicina se basan en la detección de estas mutaciones, aunque se han encontrado mutaciones en otras regiones del gen, pero con menor frecuencia.
Resistencia a Pirazinamida
La pirazinamida es un compuesto sintético redescubierto en la década de los 80 que ha facilitado el tratamiento antituberculoso de corta duración. Su mecanismo de acción no ha sido bien dilucidado, aunque se ha señalado la importancia de la acción de la enzima pirazinamidasa. Las cepas de micobacterias susceptibles a pirazinamida sintetizan pirazinamidasa, una enzima que transforma la pirazinamida en su metabolito activo, el ácido pirazinoico, el cual además de su actividad específica parece tener la capacidad de disminuir el pH del medio intracelular por debajo de los límites de tolerancia de la bacteria(35). En cepas de M. tuberculosis con resistencia adquirida y M. bovis con resistencia constitutiva a pirazinamida, se han identificado interrupciones en el gen pncA, codificante de la enzima pirazinamidasa/nicotinamidasa(35-38). Entonces, uno de los mecanismos de resistencia a pirazinamida propuestos hasta el presente es la deficiencia en pirazinamidasa, con la subsecuente pérdida de la capacidad de activar el antibiótico (Tabla 3)(37).
Resistencia a Estreptomicina
La estreptomicina es un antibiótico aminoglucósido bactericida que actúa sobre los ribosomas inhibiendo la síntesis de proteínas (Tabla 3). En la mayoría de cepas de M. tuberculosis resistentes a estreptomicina se han encontrado mutaciones en el gen rpsl que codifica la proteína ribosomal S12(18,24,39,41). Otra mutación identificada, pero de menor frecuencia se localiza en el gen rrs que codifica el ARN ribosomal 16S en una región que interactúa con la proteína S12(24,40,41).
Resistencia a Etambutol
El etambutol es un compuesto sintético que actúa como bacteriostático, cuyo mecanismo de acción es inhibir la síntesis de componentes de la pared micobacteriana a las dosis habituales. Las alteraciones génicas identificadas hasta ahora se concentran en una región designada como embCAB, que incluye genes codificantes para arabinosiltransferasas (Tabla 3), enzimas que participan en la síntesis de componentes únicos de la pared celular de micobacterias(24,41-43). Las mutaciones en la región emb se asocian a altos niveles de resistencia y se han identificado en aproximadamente el 65% de los aislados clínicos resistentes a etambutol(24,41,42).
ENSAYOS GENÉTICOS PARA DETECCIÓN DE RESISTENCIA A DROGAS
La posibilidad de un diagnóstico rápido de tuberculosis y determinación de patrón de susceptibilidad de una cepa es muy limitado por procedimientos convencionales de los laboratorios de diagnóstico microbiológico. El procedimiento estándar de diagnóstico consiste en aislar la bacteria a través del cultivo primario en medio sólido, seguido de una segunda ronda de cultivo para ensayar susceptibilidad a drogas. M tuberculosis es un organismo de crecimiento lento, con un tiempo de generación estimado en 18 a 24 horas, haciéndose las colonias visibles a las 3 a 4 semanas de incubación y en casos extremos, el aislamiento primario puede extenderse hasta 8 semanas. El tiempo mínimo promedio para el reporte definitivo de susceptibilidad a drogas, por procedimientos bacteriológicos convencionales, es de aproximadamente 45 días, pudiendo requerir hasta 3 semanas adicionales. Otra desventaja de los métodos convencionales de susceptibilidad a drogas es la inestabilidad de los componentes activos de la droga en diferentes medios de cultivo(44).
El conocimiento de las bases moleculares de la resistencia del M. tuberculosis a los antibióticos, ha llevado a estudiar más a fondo la naturaleza de las mutaciones en los diferentes genes, principalmente con fines de diagnóstico.
Actualmente existe un interés creciente en el estudio de cepas resistentes a rifampicina e isoniazida basado en evidencias de que las cepas de M. tuberculosis multirresistentes a drogas, desarrollan en primer lugar resistencia a alguno de estos bactericidas, utilizados como drogas antituberculosas de primera línea(31,33,34,45). Entonces, la identificación de cepas resistentes a alguno de estos antibióticos puede indicar la presencia de multirresistencia a drogas, orientar nuestra conducta hacia un análisis más completo del patrón de susceptibilidad de una cepa particular y tomar las medidas terapéuticas adecuadas y oportunas. El estudio de susceptibilidad permite también detectar las fuentes primarias de transmisión de la bacteria y desplegar medidas de control epidemiológico para evitar su propagación(6,15,16,20).
El enfoque genético para la detección de resistencia a rifampicina puede arrojar resultados definitivos en pocas horas o días. Debido a que las mutaciones se concentran en una región del gen rpoB, una estrategia posible para su identificación consiste en amplificar por PCR (reacción en cadena de la polimerasa) la región afectada, utilizando oligonucleótidos que delimitan dicha región (Figura 1). Una vez disponible el fragmento amplificado, la región puede ser analizada para identificar mutaciones asociadas a resistencia a rifampicina.
Estrategia genotípica para ensayar resistencia a antibióticos en M tuberculosis

El primer paso consiste en amplificar por PCR, el fragmento de ADN del gen donde se localizan mutaciones asociadas a resistencias a antibióticos. El análisis post-amplificación consiste en la detección de alteraciones en los sitios de corte por enzimas de restricción (RFLP) o alteraciones conformacionales de la molécula de ADN. La secuenciación es importante para confirmar e identificar la alteración de la secuencia de ADN.
La identificación puede hacerse directamente por secuenciación del fragmento amplificado(30,31,46,47). Este método de secuenciación directa no es factible en la mayoría de laboratorios de diagnóstico clínico, debido al alto costo de los equipos e insumos utilizados en secuenciación. Por esta razón, la detección de mutaciones se puede llevar a cabo analizando cambios conformacionales del ADN del gen en estudio, causadas por alteraciones de nucleótidos (Figura 1). Estos cambios estructurales pueden evidenciarse por alteraciones en el patrón de migración de los fragmentos de ADN amplificados al ser sometidos a electroforesis en geles de poliacrilamida(34,48,49).
El estudio de susceptibilidad a isoniazida resulta más complejo, debido a la heterogeneidad genética para el fenotipo de resistencia a este antibiótico, en el cual como se mencionó previamente, pueden estar involucrados los genes katG, inhA y aphC. Sin embargo, en el caso del gen katG, se pueden utilizar pruebas de fácil ejecución, tal como PCR-RFLP (polimorfismo en la longitud de fragmentos de restricción) para la detección de las mutaciones más frecuentes, particularmente mutaciones S315®T y R463®L(23,24), que explican la mayor proporción de mutaciones asociadas a resistencia a isoniazida. El análisis PCR-RFLP consiste en amplificar el segmento donde ocurren con mayor frecuencia las mutaciones y el fragmento de ADN amplificado (amplicón) se somete a digestión con enzimas de restricción (Figura 1)(51). La diferencia en el patrón de bandas de ADN, sometidas a electroforesis en geles de agarosa, indicará la presencia o no de mutaciones. Las mutaciones en el gen inhA se pueden investigar por análisis conformacionales(50,51).
La detección temprana de cepas resistentes a drogas de primera línea, contribuiría a un mejor manejo terapéutico del paciente, disminución del riesgo de propagación de cepas de M. tuberculosis multirresistentes a drogas y suministraría información epidemiológica valiosa para la ejecución y evaluación de los programas de control de tuberculosis.
REFERENCIAS BIBLIOGRÁFICAS
1. Raviglione MC, Snider DE, and Kochi A. Global Epidemiology of tuberculosis: morbidity and mortality of worldwide epidemic. JAMA 1995; 273: 220-226. [ Links ]
2. Dolin PJ, Raviglione MC and Kochi A. Global tuberculosis incidence and mortality during 1990-2000. Bull WHO 1994; 72: 213-220. [ Links ]
3. Murray CJ and Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet 1997; 349: 1269-1276. [ Links ]
4. Drobnieski FA, Pablos-Méndez and Raviglione MC. Epidemiology of tuberculosis in world. Semin Respir Crit Care Med 1997; 18: 419-429. [ Links ]
5. Dye C, Scheele S, Dolin P, Pathania V and Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 1999; 282: 677-686. [ Links ]
6. World Health Organization. Global tuberculosis control - surveillance, planning, financing. WHO Report 2004. WHO/HTM/TB/2004.331. [ Links ]
7. Dye C, Williams BG, Espinal MA, Raviglione MC. Erasing the worlds slow stain: strategies to beat multidrug-resistant tuberculosis. Science 2002; 15: 295: 2042-6. [ Links ]
8. World Health Organization. WHO Tuberculosis Programme: Framework for effective tuberculosis control. Geneva, Switzerland. 1994; WHO/TB/94: 179. [ Links ]
9. World Health Organization. Treatment of tuberculosis. Guidelines for national Programmes. Geneva, Switzerland. 1997; WHO/TB/97.220. [ Links ]
10. Loddenkemper R, Sagebiel D and Brendel A. Strategies against multidrug-resistant tuberculosis. Eur Repir J 2002; Suppl 36: 66s-77s. [ Links ]
11. Seung KJ, Gelmanova IE, Peremitin GG, Golubchikova VT, Pavlova VE, Sirotkina OB, Yanova GV, Strelis AK. The effect of initial drug resistance on treatment response and acquired drug resistance during standardized short-course chemotherapy for tuberculosis. Clin Infect Dis 2004; 39: 1321-1328. [ Links ]
12. World Health Organization. Guidelines for establishing DOTS-Plus Pilot Project 2000. 2000. Geneva, Switzerland. 2000; WHO/TB/2000.279. [ Links ]
13. Sterling TR, Lehmann HP, Frieden, TR. Impact of DOTS compared with DOTS-plus on multidrug resistant tuberculosis and tuberculosis deaths: decision analysis. BMJ 2003; 26: 574-574. [ Links ]
14. WHO/IUATLD. Global project on Anti-tuberculous Drug Resistance Surveillance. Anti-tuberculous drug resistance in the world. World Health Organization publication 1997. Geneva, Switzerland. WHO/TB/97.229. [ Links ]
15. Crofton J, Chaulet P, Maher D. Guidelines for the management of drug-resistant tuberculosis. Geneva, Switzerland. World Health Organization publication 1997. Geneva, Switzerland. WHO/TB 97.220. [ Links ]
16. Pablos-Mendez A, Raviglione MC, Laszlo A, Binkin N, Rieder HL, Bustreo F, Cohn DL, Lambregts-van Weezenbeek CS, Kim SJ, Chaulet P, Nunn P and World Health Organization-International Union against Tuberculosis and Lung Disease Working Group on Anti-Tuberculosis Drug Resistance Surveillance. Global surveillance for antituberculosis-drug resistance, 1994-1997. N Engl J Med 1998; 338: 1641-1649. [ Links ]
17. Ramaswamy SV and Musser JM. Molecular genetics basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuberc Lung Dis 1998; 79: 3-29. [ Links ]
18. Riska E, Jacobs WR, Allan D. Molecular determinants of drugs resistance in tuberculosis. Int J Tuberc Lung Dis 2000; 42: S4-S10. [ Links ]
19. Sharma SK and Mohan A. Multidrug-resistant tuberculosis. Indian J Med Res 2004; 120: 354-376. [ Links ]
20. Iseman MD. Tuberculosis therapy: past, present and future. Eur Respir J 2002; 20(Suppl 36): 87s-94s. [ Links ]
21. Zhang Y. Genetic Basis of isoniazid resistance of Mycobacterium tuberculosis. Res Microbiol 1993; 144: 143-149. [ Links ]
22. Zhang YT and Young D. Transformation with katG restores isoniazid-sensitivity in Mycobacterium tuberculosis isolates resistant to a range of drug concentrations. Mol Microbiol 1993; 8: 521-524. [ Links ]
23. Heym B, Alzari PM, Honoré N and Cole ST. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 1995; 15: 235-245. [ Links ]
24. Telenti A. Genetics of drug resistant tuberculosis. Thorax 1998; 53: 793-797. [ Links ]
25. Sacchettini JC and Blanchard JS. The structure and function of the isoniazid target in M. tuberculosis. Res Microbiol 1996; 147: 36-43. [ Links ]
26. Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, De Lisle G and Jacobs WR, Jr. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994; 263: 227-230. [ Links ]
27. Telenti A, Honoré N, Bernasconi C, March J, Ortega A, Heym B, Takiff HE and Cole ST. Genotypic assessment of isoniazid and rifampin resistance in Mycobacterium tuberculosis: A blind study at reference laboratory level. J Clin Microbiol 1997; 35: 719-723. [ Links ]
28. Nikolayevsky V, Brown T, Balabanova Y, Ruddy M, Fedorin I, Drobniewski F. Detection of Mutations Associated with Isoniazid and Rifampin Resistance in Mycobacterium tuberculosis Isolates from Samara Region, Russian Federation. Cli. Microbiol. 2004; 42: 4498-4502. [ Links ]
29. Wilson TM and Collin DM. aphC, a gene involved in isoniazid resistance of the Mycobacterium tuberculosis complex. Mol Microbiol 1996; 19: 1025-1034. [ Links ]
30. Telenti A, Imboden P, Marchesi F, Lowrie D, Cole ST, Colston MJ, Matter L, Schoolfer K and Bodmer T. Detection of rifampin-resistance mutations in Mycobacterium tuberculosis. Lancet 1993; 341: 647-650. [ Links ]
31. Telenti A, Imboden P, Marchesi F, Schmidheini T and Bodmer T. Direct, automated detection of rifampin-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemoter 1993; 37: 2054-2058. [ Links ]
32. Avkan Oguz V, Eroglu C, Guneri S, Yapar N, Oztop A, Sanic A, Yuce A. RpoB gene mutations in rifampin-resistant Mycobacterium tuberculosis strains isolated in the Aegean region of Turkey. J Chemother 2004; 16: 442-445. [ Links ]
33. Yue J, Zeng EL, Xie JP, Li Y, Liang L, Wang HH. Molecular mutations of rpoB gene of multidrug resistant Mycobacterium tuberculosis isolates from China. Yi Chuan Xue Bao 2004; 31:1332-1336. [ Links ]
34. Mokrousov I, Otten T, Vyshnevskiy B, Narvskaya O. Allele-Specific rpoB PCR Assays for detection of Rifampin-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemoter 2003; 47: 2231-2235. [ Links ]
35. Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nature Medicine 1996; 2: 662-667. [ Links ]
36. Scorpio A, Lindholm-Levy P, Heifets L, Gilman R, Siddiqi S, Cynamon M, Zhang Y. Characterization of pncA mutation in pyrazinamide-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemoter 1997; 41: 540-543. [ Links ]
37. Sreevatsan S, Pan X, Zhang Y, Kreiswirth BN, Musser JM. Mutations associated with pyrazinamide-resistance in pncA of Mycobacterium tuberculosis complex organisms. Antimicrob Agents Chemoter 1997; 41: 636-640. [ Links ]
38. Portugal I, Barreiro L, Moniz-Pereira J, Brum L. pncA mutations in pyrazinamide-resistant Mycobacterium tuberculosis isolates in Portugal. Antimicrob Agents Chemother. 2004; 48: 2736-2738. [ Links ]
39. Finken M, Kirschner P, Meier A, Wrede A, Bottger EC. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alteration of the ribosomal protein S12 gene and point mutation within a functional 16S ribosomal RNA pseudoknot. Mol Microbiol 1993; 9: 1239-1246. [ Links ]
40. Honoré N, Cole ST. Streptomycin resistance in micobacteria. Antimicrob Agents Chemoter 1994; 38: 238-242. [ Links ]
41. Tracevska T, Jansone I, Nodieva A, Marga O, Skenders G, Baumanis V. Characterisation of rpsL, rrs and embB mutations associated with streptomycin and ethambutol resistance in Mycobacterium tuberculosis. Res Microbiol 2004; 155: 830-834. [ Links ]
42. Telenti A, Philipp WJ, Sreevatsan S, Bernasconi C, Stockbauer KE, Wieles B, Musser JM, Jacobs WR Jr. The emb operon, a unique gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nature Medicine 1997; 3: 567-570. [ Links ]
43. Belanger AE, Besra GS, Ford ME, Mikusova K, Belisle JT, Brennan PJ, Inamine JM. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Nat Acad Sci USA 1996; 93: 11919-11924. [ Links ]
44. Shamputa IC, Rigouts L, And Portaels F. Molecular genetic methods for diagnosis and antibiotic resistance detection of mycobacteria from clinical specimens. APMIS 2004; 112: 728-752. [ Links ]
45. Hunt JM, Roberts GD, Stockman L, Felmlee TA, Persing DH. Detection of a genetic locus encoding resistance to rifampin in mycobacterial cultures and in clinical specimens. Diagn Microbiol Infect Dis 1994; 18: 219-27. [ Links ]
46. Karahan ZC, Atalay F, Uzun M, Erturan Z, Atasever M, Akar N. Sequence analysis of rpoB mutations in rifampin-resistant clinical Mycobacterium tuberculosis isolates from Turkey. Microb Drug Resist 2004; 10(4): 325-33. [ Links ]
47. Yam WC, Tam CM, Leung CC, Tong HL, Chan KH, Leung ET, Wong KC, Yew WW, Seto WH, Yuen KY, Ho PL. Direct detection of rifampin-resistant Mycobacterium tuberculosis in respiratory specimens by PCR-DNA sequencing. J Clin Microbiol 2004; 42: 4438-4443. [ Links ]
48. Mayta H, Gilman RH, Arenas F, Valencia T, Caviedes L, Montenegro SH, Ticona E, Ortiz J, Chumpitaz R, Evans CA, Williams DL. Evaluation of a PCR-Based Universal Heteroduplex Generator Assay as a Tool for Rapid Detection of Multidrug-Resistant Mycobacterium tuberculosis in Peru. J Clin Microbio. 2003; 41: 5774-5777. [ Links ]
49. Tracevska T, Jansone I, Broka L, Marga O, Baumanis V. Mutations in the rpoB and katG Genes Leading to Drug Resistance in Mycobacterium tuberculosis in Latvia. J Clin Microbiol 2002; 40: 3789-3792. [ Links ]
50. Herrera-Leon L, Molina T, Saiz P, Saez-Nieto JA, Jimenez MS. New multiplex PCR for rapid detection of isoniazid-resistant Mycobacterium tuberculosis clinical isolates. Antimicrob Agents Chemother 2005; 49:144-147. [ Links ]
51. Cockerill FR. Genetic methods for assessing antimicrobial resistance. Antimicrob Agents Chemother 1999; 43: 199-212. [ Links ]
