










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.27 n.1 Caracas jun. 2008
The mechanism of cancer involves short interfering RNA (siRNA)
Coral Wynter, Marcelo Alfonzo and Itala Lippo de Becemberg.
Sección de Biomembranas, Instituto de Medicina Experimental, Cátedra de Patología General y Fisiopatología, Facultad de Medicina, Universidad Central de Venezuela, Apdo 50587 Sabana Grande, Caracas, Venezuela.
Short running title: Cancer as a result of RNAi errors. Email: c.wynter@mailbox.uq.edu.au. Fax: 058 212 662 8877
Abstract
The recent discoveries of the RNA-mediated interference system in cells could explain all of the known features of human carcinogenesis. A novel idea, proposed here, is that the cell has the ability to recognize a mutated protein and/or mRNA. Secondly, the cell can generate its own short interfering RNA (siRNA) using an RNA polymerase to destroy mutated mRNA, even when only a single base pair in the gene has mutated. The anti-sense strand of the short RNA molecule (called sicRNA), targets the mutated mRNA of an oncogene or a tumour suppressor. During cell mitosis, the sicRNA complex can move into the nucleus to target the mutated gene. The sicRNA triggers the assembly of protein complexes leading to epigenetic modification of the promoter site of the mutant gene. In some instances, instead of methylation, the homologous DNA is degraded, leading to loss of heterozygosity. The factors controlling these two actions are unknown but the result is gene silencing or physical destruction of the mutant gene. An error in RNAi defence occurs when the sicRNA during methylation of the target gene, inadvertently interferes with the production of a miRNA specific for that tissue. This produces a change in the profile of correct proteins for that tissue. On a rare occasion, a preneoplastic stem cell will survive if miRNA interference switches on/off a gene involved in apoptosis, as well as a gene involved in cell proliferation and DNA damage surveillance. The driving force of carcinogenesis is the sequential loss of specific miRNAs.
Key words: Cáncer, siRNA, CpG methylation, RNAi, LOH, microRNA
Recibido: 20/10/2007 Aceptado: 18/11/2007
Text
Cancer has been studied over the last 30 years by the examination of hundreds, if not thousands, of different genes, with more still being discovered. Familial cancers, which develop in an individual usually before the age of 55 years, due to an inherited mutation in one allele of a key gene, have led to many important discoveries of the myriad of pathways that occur in spontaneous cancers due to old age. Each gene has a role in controlling cell division, apoptosis, and maintenance of the integrity of the genome or is located at a DNA damage checkpoint. It is considered highly significant if only 15-20% of all cancers in one organ have a mutation in one particular gene under examination. One allele of a gene could have hundreds of different mutations, sometimes concentrated in hotspots, usually involving only one base change, but the result inevitably led to a cancer. Thus the number of different oncogenic phenotypes in one tissue has been confusing, defying simplistic classifications.
The complex cellular ramifications of carcinogenesis resulting in a complete restructuring of the genome such as chromosomaldisintegration, fragmentation and multiple copies; methylation and demethylation of specific promoter sites of genes, possibly up to 1,000 changes in one cell; the disappearance of segments of genes, known as loss of heterozygosity (LOH); dedifferentiation and loss of all tissue specificity; has all been very puzzling. How did the cell survive and actually thrive with such massive changes in the nucleus? Faced with this enormous variability, and a vast amount of data that made little sense, the question as to what mechanism generated such chaos was studiously avoided, as there was no possible answer. Now the recent discoveries of RNA interference (RNAi) and the role of short interfering RNAs (siRNAs) as well as that of microRNAs (miRNAs) in controlling mRNA viability and protein translation in the cytoplasm begin to provide an answer to the cancer puzzle1,3.
Mechanism of RNAi
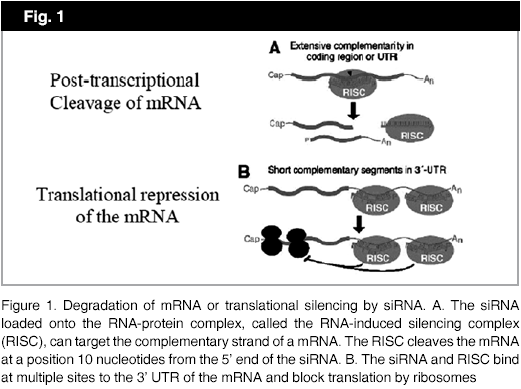
Briefly, the mechanism of RNAi is a process whereby sequence specific, post-transcriptional gene silencing can be initiated by a short dsRNA, which can be either a miRNA or a siRNA. This can happen in two ways, either by post transcriptional cleavage of mRNA through extensive complementarity in the coding region or the 3´untranslated region (UTR) or by translational repression of the mRNA again in the 3´UTR (Fig 1). Perfect homology induces mRNA degradation, but high complementarity induces repression of translation. Once the antisense strand of dsRNA or the guide strand is assembled onto a RNA-protein complex, called the RNA-induced silencing complex (RISC), it is the RISC that cleaves the targeted mRNA. Groups of mRNAs that form part of a metabolic pathway can be regulated at one time using a miRNA, which identifies it as a pleiotropic regulator of gene expression.
The process begins when a long primary miRNA transcript (pri-miRNA) is transcribed by RNA polymerase II in the nucleus. Each miRNA comes from a gene that is dedicated to the production of a particular 22-nucleotide miRNA4. At least 474 miRNAs have been identified and sequenced in the human genome as of November 20065 and probably 1500 exist which regulate 5300 genes6 although this figure could be as high as 7500 genes. [See the miRNA registry at www. microrna.sanger.ac.uk/Software/Rfam/mirna/index]. The primiRNAs are substrates for nuclear RNase III enzymes called Drosha, which is linked with another protein DGCR8 and together form a microprocessor (Fig. 2).

Following this initial processing, these molecules fold to produce hairpin structures of about 70 nucleotides, labeled precursor miRNAs (pre-miRNA), which are recognised by the two nucleotide 3´overhang (for reviews7,10). They are escorted out of the nucleus through the nuclear pore by a transport receptor, EXPORTIN5, and a Ran GTPase11. The pre-miRNAs are handed over to another RNase complex called DICER in the cytoplasm for a second processing to produce a miRNA or a siRNA duplex (Fig. 3). The only difference between a miRNA and a siRNA is that the former are normal products of the nucleus and the latter are synthetic. Generally, miRNA derive from RNA transcripts containing a stem loop and load onto the RISC as single-stranded RNA. siRNAs begin as double stranded RNA, usually depend on an artificial delivery system to the cell and require selection of the antisense strand for incorporation into the RISC12.

DICER is part of the larger protein complex that has several cofactors, a TAR RNA binding protein (TRBP), PACT and AGO213,15. TRBP recruits the DICER complex to AGO2 for miRNA processing. AGO2, a member of the family of Argonaute proteins, which are highly basic proteins of about 100 kD, contains two RNA binding domains. The PIWI domain binds the small RNA guide at its 5´end and the PAZ domainwhich binds the single stranded 3´ end of the small RNA16. In humans, AGO2 is the only one of four Argonaute proteins that can cleave targeted mRNA. It was originally thought that the rejected strand or sense strand of the dsRNA was separated by a helicase, but it has been shown that the destruction of this strand is carried out by AGO217.
Cancer and RNAi
What is the relevance of RNAi to cancer? Several dogmas abound that prevent the above mechanism from being fully utilised in our understanding of carcinogenesis. The above experiments were largely carried out in non-mammalian organisms such as plants, fungi and yeast, particularly Arabidopsis, Caernorhabditis elegans, Drosophila melanogaster, Tetrahymena thermophila and Schizosaccharomyces pombe, all with smaller genomes subject to easier genetic manipulation than mammalian cells.
The first dogma that should be re-examined is that the mammalian cell does not possess a protein checking system, that is, a system which can check the functioning or tertiary structure of the newly synthesized protein. The cell has a highly regulated system for degradation of proteins through ubiquitylation. [for review see18) The highly conserved ubiquitin protein of 76 amino acids is covalently conjugated in a regulated multistep process by a ligase to the protein to be degraded, by as many as four ubiquitin moieties. Many short lived proteins are marked for degradation by the 26S proteasome, which is composed of a 20S catalytic chamber, capped at both ends with19S regulatory units. Ubiquitin status plays a major role in protein trafficking, and acts as a sorting signal at the multivescicular body and at the plasma membrane. Ubiquitylation can also induce endocytosis. The ubiquitin pathway is a major regulator of the protein quality of misfolded proteins entering the Endoplasmic Reticulum. These misshapen proteins are marked by ubiquitin for rapid degradation in the cytoplasm by 26S proteasomes19.
To initiate a protein check and subsequent degradation, it would be expected that the errant protein must be partially inactive. Not every mutant protein or mutated mRNA elicits a carcinogenic response. The DNA repair protein, O6-methylguanine-DNA methyltransferase (MGMT) was engineered to contain eight mutations within the active site of the enzyme, although it still retained full activity. This mutated mRNA and protein was tolerated with no ill effects, at least under cell culture conditions20. It would be expected that the cell couldoperate a sophisticated system to check protein function in much the same way the cell operates multiple DNA repair mechanisms that survey the newly transcribed DNA strand to ensure fidelity.
RNA-Dependent RNA Polymerase and sicRNA
The second dogma that should be re-examined is that there is no RNA-dependent RNA polymerase (RdRP) in mammalian cells that could synthesize a short antisense RNA. However research has concentrated on finding a RNA-dependent RNA polymerase such as RNA polymerase III, which behaves like a nuclear RNA polymerase, synthesizing molecules of a large size, many thousands of base pairs. A specific search for a cytoplasmic located RdRP that synthesizes short RNAs of a length as short as 20-25 bps and no longer than 200 bps, similar to a miRNA is necessary. The cDNA of such an enzyme might be very different from the RdRP found in plants.
An important assumption of my hypothesis is that the cell in self-defence against rogue mRNAs is able to generate its own siRNA, which specifically attacks the mutated sequence to carry out degradation of the homologous mRNA. This may be initiated on a signal from the protein-checking system or from a mRNA checking system, based on a misfolded protein structure or inadequate function. In the following discussion, sicRNA here refers to endogenously generated, Small Interfering Cellular RNA, made by the cell itself, using a 22 nucleotide sense strand of the mutant section of mRNA as a guide, in response to a dysfunctional protein. This mechanism would imply that the metazoan cell has a putative RdRP that is able to direct the synthesis of a short section of antisense RNA (maybe no more than 22 nucleotides) by an unknown recognition process of the aberrant mRNA.
The resulting dsRNA consisting of the mutant, short section of mRNA and the sicRNA is processed by DICER, binds to the RISC complex, the sense strand is degraded and the sicRNA marks the mRNA for degradation (Fig.4). An RNA Polymerase IV (RNApol IV) found in plants, fungi and Caenorhabditis elegans, can generate thousands of copies of an siRNA and is the basis of quelling21. So far no RNApol IV has been found in Drosophila, insects and mammals but the evidence from the extensive data on cancer suggests that there is some sort of mechanism for generating such new sicRNAs. Perhaps in mammals, a sicRNA, using a different enzyme or another form of an RNA polymerase pol II or pol III might be involved. It is not suggested that the new sicRNA can move throughout the tissue thus providing systemic resistance as in plants.

The sicRNA may be represented by the newly discovered class of 750 tiny non-coding RNAs (tncRNAs), of 20 nt, discovered in C. elegans22. They are precisely complementary to mRNA from more than 500 different genes and seem to be endogenous siRNAs. So far these tncRNAs have not been found in mammals. It is clear the defence system must be infinitely adaptable as in any one oncogene hundreds of different mutations have been identified. For example, there are 300 known mutations in the APC gene; there are over 2000 different mutations in the two breast cancer genes BRCA1 and BRCA2; and 225 different mutations in the DNA mismatch repair gene, MLH1, have been sequenced. Usually only one organ of the body is targeted for malignancy in a familial cancer, although every cell in all tissues carries the mutation. Hence there must be a general, efficacious, adaptive mechanism producing sicRNAs for targeting thousands of differently mutated mRNAs that can arise. Otherwise, in an inherited cancer every cell in the body would be clogged with large amounts of aberrant mRNAs and dysfunctional proteins and every tissue would produce a cancer.
Transport of sic RNAs into the nucleus
Another key postulate of this theory is that in a cancer-prone tissue, the anti-sense strand sicRNA made against the mutant mRNA in the cytoplasm is able to be transported back into the nucleus during cell division and initiate one of three outcomes, hypermethylation or hypomethylation of the promoter site of the mutant gene or LOH of the gene itself. It is thought the siRNA enters the nucleus during cell division when the nuclear membrane breaks down. The factors determining which one of these three results occur are not known. The RISC complex is not found in the nucleus, so a different protein complex binds to the sicRNA to cause its translocation into the nucleus and activation of the methylating machinery. There is some evidence that an siRNA bound to the protein complex of an RNA-Induced Initiation of Transcription gene Silencing (RITS) can carry out methylation. The sicRNA binds to the homologous DNA being transcribed by RNA polymerase II and methylation of the promoter site ensues, switching off the mutant gene. An illustration of these processes is shown in figura 4.
Another key postulate of our theory is that the methylation or LOH of an oncogene or tumour suppressor due to the action of a sicRNA interferes with the expression of an miRNA, fundamental to that particular tissue. The sicRNA is generated to cope with any mutation, and once linked up with the RITS machinery, it can affect CpG methylation of the targeted gene.
It should be noted that CpG methylation can also switch on gene expression. Thus the sicRNA of a particular oncogene could theoretically bind to sites all over the genome by annealing to 6-8 nucleotides on an homologous DNA strand, but this would produce a cancer in many different tissues, depending on the sequence of the mutated site. As this does not happen and a mutation of an oncogene always produces a cancer in one specific tissue, the answer must be linked to the production of specific miRNAs for that particular tissue. For example, an inherited mutation in the MLH1 gene always initiates a cancer in the large colon, and sometimes in ovarian tissue, but these are derived from the same embryonic tissue, but not in the lung nor liver nor prostate gland, even though the mutation is present on one allele in every cell in the body. An examination of what is currently known of miRNAs supports this argument.
The exact consequences of such interference with a miRNA is not known. A sicRNA synthesized by the cell to stop translation of a rogue mRNA, enters the nucleus during mitosis. The sicRNA attached to a protein complex, binds to a homologous region of the gene, methylates the promoter site and inadvertently prevents the production of a miRNA close to that site or within the site. In a slightly different scenario, some molecules of the sicRNA binds to an miRNA undergoing transcription and abrogates its action. In an attempt to silence the production of the wrong genes for that tissue, other genes are silenced by the same sicRNA mechanism. This could result in hundreds of genes being methylated and silenced and more miRNAs being disrupted. All of these events must occur in the stem cell.
Most cells with a disabled miRNA will be eliminated by apoptosis. Thus for the stem cell to survive, two processes must be deactivated, apoptosis and normal control of cell proliferation. In another scenario, the sicRNAs may compete with a small modulatory dsRNA (smRNA), such as found in the neuron, described in detail below23, and switch off synthesis of a large group of tissue specific mRNAs. The type of miRNA that is disabled may depend on it being actively transcribed at the same time the sicRNA enters the nucleus. In the next round of cell division, a different miRNA could be inactivated, producing a cancer cell with a number of different characteristics. It may need several rounds of cell division before a second miRNA is disabled. This would add enormous variation to the resulting phenotype, as described above with different siRNAs against p53. Hence the cell begins a disintegration and a dedifferentiation of the particular architecture of that tissue. The disruption of a specific miRNA marks a qualitative step that signals the progress from a normal stem cell to a harmless Aberrant Crypt Foci (ACF), to an adenoma or a hyperplastic polyp, to a serrated adenoma, at least in colon cancer24.
The hypotheses of this model that need to be proven experimentally in humans are:
1 There is a protein/mRNA checking system for aberrations, perhaps based on poly A, similar to tRNA in yeast or a system, assessed by the number of ubiquitylation and deubiquitylation sites which can correctly diagnose a misfolded protein.
2 The cell can generate its own siRNAs by use of a mammalian RNA-dependant RNA polymerase with a sequence that is the anti-sense strand to bind to any short section (20–25 nt) of mutated mRNA that may arise.
3 The newly synthesized siRNA, called sicRNA, can hinder translation or degrade the mutated mRNA through the RISC complex.
4 In all tissues, the sicRNA/RITS complex can move into the nucleus during cell division and silence the specific mutated gene by CpG methylation or LOH. In non-cancerous tissue, which also carries the mutant allele, this is the end of the story.
5 A pre-tumor begins when CpG Methylation and/or LOH disrupt a key miRNA specific for that tissue. For the stem cell to survive, at least two processes, one involved in cell proliferation and the other in apoptosis, must be disabled.
6 A qualitative leap in pathogenesis occurs when a sicRNA abrogates a pleiotropic miRNA, essential for that tissue, leading to cell dedifferentiation.
7 Cancers can be initiated by many different pathways, utilizing sicRNAs, either elicited by a familial germline mutation or by a spontaneous mutation which is deleterious to that tissue, because of the specific miRNA. The exact phenotype even in the same individual will depend on the initial error and the order of subsequent random events.
Acknowledgement
The authors are indebted to CDCH PI.09.6030.2005 to Dr. Itala Lippo de Becemberg to support Dr. Coral Wynter in Venezuela.
References
1. Filipowicz W. RNAi: the nuts and bolts of the RISC machine. Cell 2005; 122: 17-20. [ Links ]
2. Hall PA, Russell SH. New perspectives on neoplasia and the RNA world. Hematol Oncol 2005; 23: 49-53. [ Links ]
3. Wynter CVA. The dialectics of cancer: A theory of the initiation and development of cancer through errors in RNAi. Medical Hypotheses 2006; 66: 612-635. [ Links ]
4. Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science 2003; 299: 1540. [ Links ]
5. Griffith-Jones S. The Micro RNA Registry, NAR 2004; 32: Database Issue D109-D111 [ Links ]
6. Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science 2005; 309: 1519-24. [ Links ]
7. Ambros V. The functions of animal microRNAs. Nature 2004; 431: 350-5. [ Links ]
8. Karagiannis T, EL-Osta A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther 2005; 12: 787-95. [ Links ]
9. Mattick J, Makunin IV. Small regulatory RNAs in mammals. Hum Mol Genet Apr15 2005; 14: R121-32. [ Links ]
10. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 2004; 5: 522-31. [ Links ]
11. Kim VN. MicroRNA precursors in motion: exportin-5mediates their nuclear export. Trends in Cell Biol 2004; 14: 156-9. [ Links ]
12. Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006; 20: 512-24. [ Links ]
13. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005; 436: 740-4. [ Links ]
14. Haase AD, Jaskiewicz L, Zhang H, Laine S, Sack R, Gatignol A, Filipowicz W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Reports 2005;6:961-7. [ Links ]
15. Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. EMBO Journal 2006; 25: 522-32. [ Links ]
16. Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nature Cell Biol 2005; 7: 719-23. [ Links ]
17. Matranga C, Tomari Y, Shin C, Bartel DP, Zamore PD. Passengerstrand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005; 123: 607-20. [ Links ]
18. Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Develop Biol 2006; 22: 159-80. [ Links ]
19. Meusser B, Hirsch C, Jaroosch E, Sommer T. ERAD: the long road to destruction. Nature Cell Biol 2005; 7: 766-72. [ Links ]
20. Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci USA 2003; 100: 766-81. [ Links ]
21. Mello CC, Conte D. Revealing the world of RNA interference. Nature 2004; 431: 338-42. [ Links ]
22. Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol 2003; 13: 807-18. [ Links ]
23. Kuwabara T, Hsieh J, Nakashima K, Taira K, Gage FH. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 2004; 116:779-93. [ Links ]
24. Wynter CVA, Walsh MD, Higuchi T, Leggett BA, Young J, Jass JR. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Gut 2004; 53: 573-80. [ Links ]
