










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.28 n.1 Caracas ene. 2009
Mecanismos del dolor neuropático: Del laboratorio a la clínica.
1,2Juan Vicente Gómez-Barrios y 1Víctor Tortorici
1 Laboratorio de Fisiopatología del Sistema Nervioso. Centro de Medicina Experimental. Instituto Venezolano de Investigaciones Científicas (IVIC). Apartado 20632, Caracas 1020A, Venezuela.
2 Postgrado de Farmacología. Facultad de Farmacia. Universidad Central de Venezuela. Caracas 1020A, Venezuela. J.V. Gómez-Barrios. Doctor en Farmacología V. Tortorici. Doctor en Biofísica y Bioquímica Correspondencia: Juan V. Gómez Barrios. Postgrado de Farmacología. Facultad de Farmacia. Decanato. Universidad Central de Venezuela. Ciudad Universitaria. Caracas 1020A, Venezuela. Teléfono: +58-212-202.21.69 e-mail: jvgomezbarrios@gmail.com
Resumen
El dolor neuropático es un síndrome de muy difícil manejo en la práctica clínica. Suele originarse a consecuencia de lesiones o enfermedades que afectan al sistema somatosensorial. Lesiones de nervios periféricos, la diabetes, el virus herpes zoster, entre otras, son ejemplos de entidades clínicas que causan dolor neuropático. Este tipo de dolor supone modificaciones de la fisiología normal de las neuronas que integran la vía de transmisión nociceptiva y que operan tanto a nivel periférico, como central. Entre estos mecanismos se incluye la generación de descargas ectópicas, cambios en el genoma de las neuronas involucradas, alteración de canales iónicos, pérdida de las actividades inhibitorias endógenas, activación anormal del sistema inmunitario y sensibilización tanto periférica, como central. Este artículo resume los mecanismos moleculares y neuroplásticos que ocurren en situaciones de dolor neuropático y revisa diferentes modelos experimentales para su estudio. El desarrollo de estos modelos ha permitido dilucidar los mecanismos subyacentes de esta patología, el diseño de nuevas estrategias terapéuticas y el diagnóstico acertado de la enfermedad.
Palabras claves: modelos experimentales, sensibilización, NMDA, neuropatía, sistema inmunitario, daño a nervios.
Abstract
Neuropathic pain is a very complex clinical syndrome that results as a consequence of lesions or diseases affecting the somatosensory system. Lesion of peripheral nerves, diabetes, herpes zoster virus, among others, are clinical entities that may cause neuropathic pain. Neuropathic pain also reflects changes of the normal physiology of the nociceptive transmission neurons that may happen at peripheral and central levels. These mechanisms include the generation of ectopic discharges, genomic changes of the neurons involved, alterations of ionic channels and of the endogenous inhibition of pain, abnormal activation of the immune system as well as peripheral and central sensitization. This article review the molecular and neuroplastic mechanisms associated to neuropathic pain and discuss different experimental models for its study. The development of these models has been helpful to understand the subjacent mechanisms of this pathology, and has contributed to design new therapeutical approaches and the correct diagnose of the disease.
Key words: experimental models, sensitization, NMDA, neuropathy, immune system, nerve lesions.
Recibido: 13/12/2008 Aceptado: 04/03/2009
Dolor neuropático
La Asociación Internacional para el Estudio del Dolor (International Association for the Study of Pain, IASP), define al dolor neuropático como un dolor iniciado o causado por una lesión primaria o disfunción en el sistema nervioso periférico o en el sistema nervioso central (Merskey y Bogduk, 1994). Más recientemente, el Grupo de Interés Especial sobre Dolor Neuropático de la IASP [Special Interest Group on Neuropathic Pain (NeupSIG)] lo define como el dolor causado por lesión directa o enfermedad que afecta al sistema somatosensorial (Treede y col., 2008). En esta definición la palabra enfermedad reemplaza a disfunción, con la intención de referirse a procesos patológicos específicos, como por ejemplo, inflamación, condiciones autoinmunes o canalopatías; mientras que la palabra lesión, se refiere a un daño micro-o macroscópicamente identificable (Gómez-Barrios, 2007). En este caso el dolor deja de ser una señal fisiológica asociada a la búsqueda de protección, perdiendo su condición adaptativa, para convertirse en un estado patológico, que involucra una serie de elementos que facilitan su generación y persistencia en el tiempo. Son ejemplos de dolor neuropático la neuralgia del trigémino (Zakrzewska y Patsalos, 2002), la neuropatía diabética (Wilson, 1999; Wolsteind y col., 2005), la neuralgia post-herpética (Mielki y col., 2005; Sabadowsk y col., 2004), las monorradiculopatías (Jensen y col., 2007), el dolor neuropático inducido por radiación y quimioterapia (Caraceni y col., 2004; Ragajopal y col., 2004; Staats y col. 2004), el dolor de miembro fantasma (Huse y col., 2001; Smith y col., 2005), y el llamado síndrome doloroso regional complejo (Weber, 2004), entre otros.
El dolor neuropático presenta una variedad de síntomas y puede percibirse como una sensación quemante, persistente y/o lancinante (Chong y Bajwa, 2003), que frecuentemente está asociado a signos sensoriales como la alodinia (dolor que resulta al aplicar un estímulo inocuo), o la hiperalgesia (incremento de la respuesta a un estímulo que normalmente es doloroso) (Baron, 2006; Cruciani y Nieto, 2006; IASP, 1994). También pueden observarse la presencia de signos sensoriales anormales de tipo espontáneo o evocado, tales como disestesias (sensaciones anormales y displacenteras) y parestesias (sensaciones anómalas no desagradables) (Attal y Bouhassira, 1999; Baron, 2006).
Modelos experimentales para el estudio del dolor neuropatico
Son diversos los tipos de neuropatias que estan distinguidas en la clinica (Campbell y Meyer, 2006). Sin embargo, se sugiere, pero no se ha demostrado en su totalidad, que al menos todos los tipos de dolor neuropatico comparten un mecanismo fisiopatologico comun (Besson, 1999; Bridges y col., 2001; Zimmermann, 2001). Con el objeto de estudiar esta patologia se han desarrollado diversos modelos de experimentacion. Estos generalmente consisten en: a) lesiones parciales o totales del nervio ciatico, b) lesiones de nervios raquideos en la region lumbar, y c) utilizacion de farmacos como el placlitaxel (Polomano y col., 2001), la vincristina (Autier y col., 1999; Siau y col., 2006) y la estreptozotocina (Bennett, 2001; Calcutt y Chaplan, 1997). Estos ultimos producen dolor neuropatico por induccion de toxicidad en los nervios perifericos.
En la mayoria de los modelos que se utilizan en la actualidad, las alteraciones son realizadas en un miembro posterior, causando un dano parcial en los nervios perifericos o espinales (Bennett, 2001; Bridges y col., 2001). En este tipo de modelos tipicamente se estudian la hiperalgesia y la alodinia inducidas mediante la aplicacion de estimulos termicos o mecanicos, que son los mas utilizados para determinar el grado de nocicepcion en los animales de experimentacion. Entre los modelos mas empleados para generar un trauma mecanico en los nervios figuran la constriccion cronica del nervio (CCN) (Bennett y Xie, 1988), el modelo de ligadura parcial del nervio ciatico (LPN) (Seltzer y col., 1990), el modelo de ligadura del nervio espinal (LNS) (Kim y Chung, 1992) y el modelo de ligadura neural por omision (LNO) (Decosterd y Woolf, 2000).
El modelo de CCN consiste en realizar cuatro ligaduras laxas en el nervio ciatico antes de su trifurcacion en la zona poplitea. Al ser laxas, las ligaduras impiden, pero no bloquean totalmente, la circulacion en la porcion del nervio que resulta afectada. En respuesta a las ligaduras se desarrolla una reaccion inflamatoria que conlleva a la perdida de la mayoria de fibras A (,) y a una leve reduccion de las fibras C (Basbaum y col., 1991; Sugimoto y col., 1990; Tandrup y col., 2000). Con este procedimiento los signos sensoriales de alodinia e hiperalgesia son medidos exitosamente (Hamidi y col., 2006). Ademas, el modelo de CCN permite evaluar al animal en ambos miembros posteriores para efecto de comparacion, manteniendose incluso la presencia de reflejos nocidefensivos en la pata afectada (Bennett, 2001; Bridges y col., 2001; Joshi y col., 2006).
El modelo de LPN consiste en una ligadura fuertemente ajustada alrededor de una porcion del nervio ciatico, la cual produce una desaferentacion parcial, pero no diferencial.
De hecho, con esta maniobra alrededor de 2/3 de la poblacion total de fibras resulta eliminada (Begon y col., 2002; Bennett, 2001). Por otro lado, este modelo proporciona un menor grado de inflamacion, en comparacion al modelo de CCN, pero produce dolor espontaneo. Este modelo, al igual que el de CCN, permite evaluar al animal en ambos miembros posteriores para efecto de comparacion (Kim y col., 1997; Seltzer y col., 1990).
El modelo de LNS consiste en producir un dano en los nervios espinales L5 y L6, por medio de una ligadura realizada con un alto grado de ajuste, lo cual provoca una seccion transversal del nervio (Kim y Chung, 1990). En este modelo, los miembros posteriores dejan de ser inervados en mas de un 50%, afectando incluso a los ganglios vecinos de la raiz dorsal (Li y col., 2000). Este modelo es significativamente mas invasivo que el de CCN e induce conductas nociceptivas por un tiempo mas prolongado (Bridges y col., 2001; Xiao y col., 1994).
Mas recientemente, Decosterd y Woolf (2002), desarrollaron el modelo LNO, que consiste en realizar una seccion transversal, por separado, en los nervios peroneal y tibial, dejando intacto el sural del miembro posterior. De esta forma, el modelo permite evaluar territorios de la piel no danados y territorios de areas desnervadas. Este modelo produce una rapida y prolongada (>6 meses) modificacion conductual (nocicepcion). Por otro lado, empleando variaciones de este metodo de lesion en los que se lesiona cada rama del nervio se expresan con mayor facilidad algunos signos de neuropatia, tales como alodinia mecanica, alodinia por frio y dolor espontaneo (Lee y col., 2000; Shields y col., 2003).
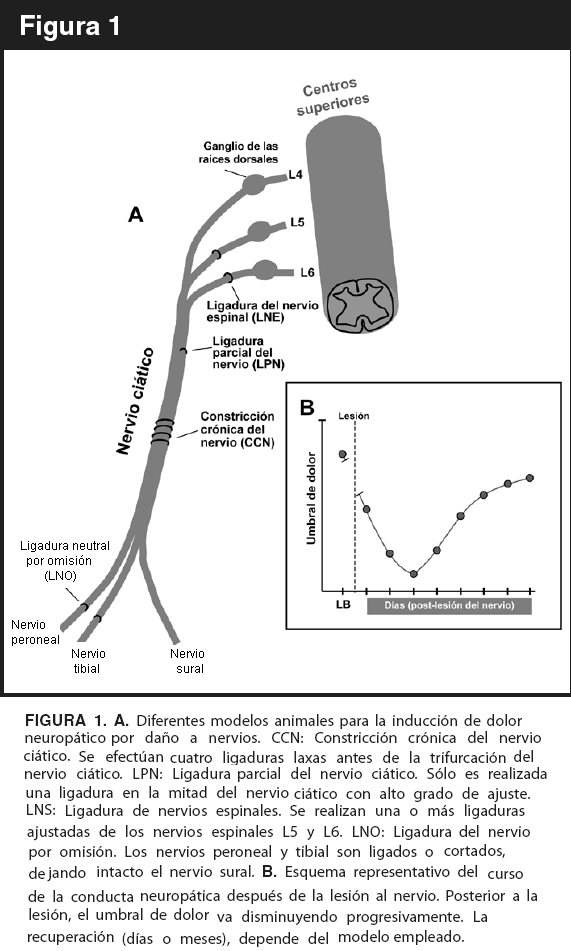
Los modelos anteriormente descritos producen signos conductuales característicos del dolor neuropático, que también son observados en los pacientes con este tipo de patología (Bennett, 2001; Kim y col., 1997). El dolor anormal que es producido en los modelos de LPN y LNO aparece luego de las primeras horas posteriores a la cirugía, a diferencia del modelo de CCN, en el que el dolor puede aparecer a partir de cuarto día posterior a la lesión (Kim y Chung, 1991; Seltzer y col., 1990). Al emplear los modelos de LPN y LNO es posible observar las conductas nociceptivas a lo largo de dos meses, a diferencia del modelo de LNS, que puede perdurar hasta los seis meses, o del modelo de CCN, en el que pueden persistir alrededor de dos o tres semanas postcirugía (Bridges y col., 2001; Erichsen y Blackburn-Munro, 2002).
Mecanismos periféricos y centrales generadores del dolor neuropático
Los mecanismos responsables de la aparición del dolor neuropático se clasifican en periféricos y centrales. Los mecanismos periféricos implican, entre otros, la generación de una actividad espontánea anormal (descargas ectópicas) en los aferentes primarios, la disminución del umbral de activación de los nociceptores (Bridges y col., 2001), la comunicación cruzada entre fibras de transmisión (Amir y Devor, 2000), la sobreactividad de los canales de sodio en los nervios periféricos (Omana-Zapata y col., 1997) y la inflamación del nervio afectado (Attal y Bouhassira, 1999; Baron, 2000; Campbell y Meyer, 2006). El daño de las neuronas sensoriales puede desarrollar cambios en la excitabilidad de las neuronas vecinas, aún en las que no resultan inicialmente afectadas por la lesión, y estos cambios pueden generar potenciales de acción al aplicar cualquier tipo de estímulación en la periferia, inclusive estimulación inocua (alodinia). Estos cambios pueden manifestarse a lo largo de la vía de transmisión nociceptiva
Las descargas ectópicas son más frecuentes en las fibras A; sin embargo, también ocurren en un grado más limitado en fibras o axones desmielinizados (Liu y col., 2002; Woolf, 2004). Al producirse un foco ectópico, la frecuencia de impulsos que se transmite a lo largo de la vía del dolor puede ir en aumento en la medida en que la información avanza hacia su sitio de procesamiento final en la corteza cerebral. Esto significa que cada estación de relevo podría actuar como un amplificador de señales, deformando totalmente el mensaje nociceptivo (Tortorici, 2008). Entre los factores que aparecen como responsables de las descargas ectópicas figuran la sensibilización (up-regulation) de los canales de sodio dependientes de voltaje (incluyendo el NaV1.3, NaV1.7 y NaV1.8), la desensibilización (down-regulation) de los canales de potasio, y la posible reducción del umbral de los canales receptores transitorios de potencial (Trp), que aunque son sensibles a cambios de temperatura también responden al tacto y al dolor (Waxman y col., 1999).
La actividad ectopica puede generar parestesia, disestesia y dolor de tipo quemante (Baron, 2006). Por ejemplo, se sugiere que la actividad espontanea de los nociceptores de tipo C, es responsable de la sensibilizacion de las neuronas del asta dorsal (Woolf y Mannion, 1999) y que la actividad espontanea de las fibras A mielinizadas (las cuales transmiten senales no-nocivas), esta relacionada inicialmente con parestesias, pero posteriormente con disestesias y dolor (Woolf, 2004).
Despues de un dano a un nervio periferico no solo los canales de Na+ estan alterados. Tambien los canales de calcio participan en la generacion de la alodinia y la hiperalgesia (Aurilio y col., 2008). El uso de antagonistas especificos de los canales de Ca+2 tipo N (neuronales), produce una reduccion de la hiperalgesia termica y la alodinia mecanica en animales con CCN, cuando son administrados directamente en el sitio de la lesion (Bridges y col., 2001; Xiao y Bennett, 1995). Otros estudios han demostrado que la administracion subcutanea de antagonistas de estos canales atenua la hiperalgesia termica y la mecanica inducida por LPN, sugiriendo el efecto local de estos canales en la generacion de hiperalgesia (White y Cousins, 1998). Un hallazgo reciente ha descrito que en la neuropatia inducida por CCN se incrementa la expresion de los canales de Ca+2 tipo T en neuronas del ganglio de la raiz dorsal (Jagodic y col., 2008).
Con respecto a los mecanismos centrales, ocurren diversas variaciones neuroquimicas en el entorno de la lesion, tales como la liberacion de glutamato (Anbar y Gratt, 1997), de sustancia P, de oxido nitrico (Bardoni y col., 2004; Morisette y Nagy, 1999), e incluso modificaciones de la citoarquitectura neuronal (neuroplasticidad), que podrian inducir la activacion patologica de las neuronas nociceptivas centrales (Attal y Bouhassira, 1999; Woolf y Salter, 2000).Tambien se considera anormales la disminucion del umbral de activacion de las neuronas de relevo de la via del dolor (Coderre y col., 1993; Nakamura y Atsuta, 2004), y las alteraciones del sistema de modulacion endogena del dolor (McHugh y McHugh, 2000).
El dano a los aferentes primarios en los nervios perifericos puede inducir severos cambios anatomicos en el asta dorsal de la medula espinal (Coderre y col., 1993; Woolf, 2004). En condiciones fisiologicas normales, las diferentes fibras de los aferentes primarios terminan en zonas especificas del asta dorsal denominadas laminas (Rexed, 1954). Por ejemplo, las fibras A y las fibras C, normalmente asociadas a la transmision nociceptiva, alcanzan las laminas I, II y V, mientras que las neuronas de las fibras A (tipicamente asociadas al tacto) terminan en las laminas III y IV (Baron, 2006; Woolf y Salter, 2000). Sin embargo, esta distribucion especifica puede verse afectada bajo ciertas condiciones patologicas, incluyendo el dolor neuropatico (Polgar, 2004). Estas reorganizaciones neuronales son causadas por la expresion de diversos factores neurotroficos y podrian provocar que estimulos inocuos puedan percibirse como dolorosos (alodinia) (Tandrup y col., 2000; Wright, 1999). Asi, en animales de experimentacion sometidos a una axotomia del nervio ciatico, las fibras A logran alcanzar las laminas superficiales del asta dorsal donde se encuentran las neuronas nociceptivas y de alli proyectan hacia la corteza somatosensorial, en las areas de procesamiento nociceptivo, de forma que un estimulo inocuo podria ser percibido como un estimulo nocivo y asi generar dolor. Esto ocurre en la primera semana posterior al dano del nervio y puede extenderse hasta seis meses despues de la cirugia (Woolf y col., 1995). La neuroplasticidad tambien ha sido estudiada directamente en humanos. Estudios electrofisiológicos efectuados durante una talotomía, han demostrado una reorganización profunda de esta estructura en pacientes con dolor neuropático, en comparación con pacientes que sólo presentan desórdenes motores (Lenz y col., 1998).
Los incrementos en la excitabilidad de las neuronas de la vía del dolor que se producen en condiciones de neuropatía son consecuencia directa del fenómeno conocido como sensibilización (Woolf y Mannion, 1999; Woolf y Ma, 2007). Bajo esta condición ocurre una disminución del umbral de respuesta de las neuronas nociceptivas (Besson, 1999), que a su vez trae como consecuencia un incremento en la generación y en la frecuencia de transmisión de los impulsos en la vía del dolor. Dependiendo del lugar en el que se produzca la sensibilización, esta puede ser periférica o central (Costigan y Woolf, 2000; Zimmermann, 2001).

La sensibilización es debida, al menos en parte, a la exposición a mediadores pronociceptivos en la zona de daño (Baron, 2000; Basbaum y Jessell, 2001). Por ejemplo, en estudios experimentales en ratas, se ha determinado un incremento de la actividad de la óxido nítrico sintasa, y es de suponer que también del óxido nítrico (ON), en modelos de dolor neuropático (McHugh y McHugh, 2000), alcanzándose un efecto antinociceptivo con inhibidores de esta enzima (Mayer y col., 1999; Tasorelli y col., 2006). Recientemente Naik y col. (2006), demostraron en animales neuropáticos que el uso de precursores del ON (L-arginina, nitroprusiato de sodio, etc.), potenciaban la hiperalgesia y la alodinia en ratas.
Por otra parte, la presencia de otro neuromodulador, la sustancia P, ha sido evaluada en estados de dolor crónico en ratas (Kessler y col., 1992, Nakatzuka y col., 2005), y se ha determinado su incremento en animales con neuropatías por ligadura de nervios (Liu y col., 1997). De hecho, la utilización de antisuero de sustancia P en ratas neuropáticas produce un efecto antialodínico en los animales tratados (Wu y col., 2005). En este mismo contexto, también se ha demostrado el incremento del neurotransmisor excitatorio glutamato (Karlsson y col., 2002), el de la ciclooxigenasa-2 (COX-2) (Broom y col., 2004) y el de diversos tipos de prostaglandinas (Kroin y col., 2006) en animales con neuropatía periférica. Según las evidencias presentes en la literatura, las COXs y sus productos, las prostaglandinas, resultan primordiales en el inicio del proceso inflamatorio asociado a la neuropatía, pero no en su mantenimiento (Heweet y col., 2006; Ma y Eisenach, 2003; Tasorelli y col., 2006; Vázquez y col., 2001). No obstante, existen evidencias que indican que los niveles de COX-2 se incrementan significativamente en la médula espinal luego de efectuar ligaduras alrededor de nervios espinales (Broom y col., 2004). Ese nivel de la enzima disminuye con la administración de inhibidores de la COX-2. Por otro lado, en ratas con ligadura de nervio espinal (L5), se demostró que los niveles COX-2 se incrementan tanto en el asta dorsal, como en el tálamo, de 3 a14 días después de la lesión, sin que ocurran cambios mayores en la expresión de la COX-1 (Zhao y col., 2000). Sin embargo, en el tratamiento del dolor neuropático la manipulación de ambas isoformas y su utilización como blanco terapéutico sigue siendo controversial.
Las sustancias pronociceptivas antes mencionadas modifican la capacidad de respuesta de las terminaciones nerviosas libres y de los aferentes primarios, estimulando a la membrana del nociceptor y facilitando la transducción del estímulo nociceptivo (sensibilización periférica) (Dickenson, 1996; Katz y Gold, 2006; Woolf y Salter, 2000). Si la exposición a estos cambios en el microambiente es breve (por ejemplo en el dolor agudo), se producirá una percepción dolorosa que también será breve (Basbaum, 1999; Clancy, 1995), pero si el cambio se extiende tanto en el tiempo, como en el espacio, se producirá de forma mantenida una reducción de los umbrales de activación del nociceptor y un incremento en la transmisión de los impulsos nociceptivos a lo largo del aferente primario (Costigan y Woolf, 2000; Sun y col., 2005). En consecuencia, el incremento de los impulsos nociceptivos que provienen de la periferia termina por afectar a las neuronas de procesamiento nociceptivo ubicadas en las diferentes láminas del asta dorsal, dando como resultado el fenómeno de sensibilización central (Baron, 2006; Besson, 1999; Bereiter y Bennetti, 1996). La coliberación de glutamato y de sustancia P en los terminales centrales de los aferentes primarios en las neuronas nociceptivas (Attal y Bouhassira, 1999) activa sólo a un número restringido de receptores de aminoácidos excitatorios pre- y postsinápticos (Bardoni y col., 2004; Dray y col., 2000; Lee y col., 2002). Al continuar la generación de estos impulsos nociceptivos, se logran activar otros tipos de receptores, incluyendo el denominado N-metil-D-aspartato (NMDA), tanto a nivel precomo postsináptico (Bardoni y col., 2004; Dickenson y col., 1997). Lo anterior origina impulsos anormales persistentes en la médula espinal y con ello se provoca un estado de hiperexcitabilidad y de dolor sostenido (Sun y col., 2005; Woolf y Ma, 2007).
En condiciones fisiológicas normales, la liberación de glutamato inicialmente sólo provoca la activación de los receptores de AMPA/kainato (Dickenson y col., 1997; Fields, 1987; Qian y Johnson, 2002), dado que a ese valor de corriente de membrana los receptores de NMDA están desactivados debido a que el ión magnesio se encuentra bloqueando el canal (Diglendine, 1999). Si la estimulación nociva persiste, el efecto acumulado de la liberación de glutamato produce un nivel suficiente de despolarización de la membrana postsináptica, que a su vez induce un cambio conformacional en el receptor, lo cual junto a la repulsión electrostática (producto del influjo de cationes) permite eliminar el bloqueo ejercido por el magnesio (Antonov y Johnson, 1999; Qian y col., 2002). De esta manera, el magnesio sale al exterior de la célula en conjunto con el ión potasio (Kandel y Siegelbaum, 2001). Así, el canal del receptor NMDA queda completamente desbloqueado, permitiendo el influjo iónico de calcio y sodio, lo cual se traduce en una despolarización neuronal masiva, que se añade a la ya existente (Bridges y col., 2001; Karlsson y col., 2002; Ruppersberg y col., 1994).
El calcio que ingresa a la célula por el canal del receptor de NMDA, sumado a la actividad de los canales de calcio dependientes de voltaje, activa a una serie de neuromoduladores tales como la fosfolipasa A2 (Costijan y Woolf, 2000), a diversos tipos de prostaglandinas (Tasorelli y col., 2006; Woolf y Thompson, 1991), a la sustancia P (Nakasutka y col., 2005) y a la colecistoquinina (CCK) (Ma y col., 2003; Schafer y col., 1998), entre otras. Estas sustancias, al interactuar con sus receptores postsinápticos, mantienen una prolongada excitabilidad neuronal, la cual puede ser revertida con la administración de sus respectivos antagonistas (André y col., 2005; Kandel y Siegelbaum, 2001). En este contexto, en ratas neuropáticas se ha determinado que el aumento en las concentraciones de Ca+2 en el asta dorsal (láminas I y II), disminuye después de administrarse MK-801 (un antagonista de los receptores de NMDA) en los animales de experimentación (Skyba, 2005).

Los estados de hiperalgesia primaria, los de alodinia, así como los aumentos en la duración de la respuesta frente a una estimulación breve y la hiperalgesia secundaria en los tejidos no lesionados (dolor referido) (Bridges y col., 2001), son procesos patológicos que persisten después de que ha desaparecido la lesión periférica y que al parecer dependen, en buena medida, de los cambios centrales asociados a los receptores NMDA (Costigan y Woolf, 2000). En animales con ligadura parcial de nervio ciático se demostró, por medio de histoquímica, un incremento en la expresión del receptor NMDA, específicamente de las subunidades NR1 y NR2B en el lado ipsilateral a la ligadura (Ultenius y col., 2006). El incremento en la expresión de este receptor, puede prevenirse con la administración de memantina (un antagonista de los receptores de NMDA). Por otro parte, posterior a una neuropatía periférica por CCN en ratas, se detectó un incremento de glutamato en el asta dorsal, en comparación con el grupo control (Kawamata y Omote, 1996). Cabe destacar, que ha sido demostrado que el número de receptores de NMDA se incrementa significativamente en el asta dorsal al emplear modelos de diabetes (Tomiyama y col., 2005), lo cual quizás pueda contribuir a explicar la neuropatía en los pacientes afectados.
En otro contexto, en un modelo experimental de polineuropatía inducida por ingesta de etanol, los animales expuestos a este tratamiento presentaron hiperalgesia mecánica a las 14 semanas del estudio, siendo atenuada con la administración i.p. de ifenprodil, un inhibidor selectivo de la subunidad NR2B del receptor de NMDA (Narita y col., 2007).

Dolor neuropático y el sistema inmunitario
Cuando fueron desarrollados los primeros modelos animales para inducir neuropatía, el daño al tejido y la pérdida de axones mielinizados y amielínicos en el nervio ciático fueron considerados el factor fundamental para la producción de los síntomas del dolor neuropático (Basbaum y col., 1991; Moalen y Tracey, 2006). Sin embargo, el proceso fisiopatológico asociado al dolor neuropático no sólo involucra vías neuronales, sino también a las células de Schwann, a las células satélite en el ganglio de la raíz dorsal, a la microglía, los astrocitos y a los componentes periféricos y centrales del sistema inmunitario (Campell y Mayer, 2006; Marchand y col., 2005) lo cual contribuye a facilitar la degeneración walleriana de la fibras nerviosas lesionadas (Fischer y col., 2008; Jander y col., 1996; Sholtz y Woolf, 2007; Streit, 2002). Esto significa que el dano local se extiende y conlleva a fenomenos de hiperalgesia primaria y secundaria.
Se ha descrito que inmediatamente despues de una lesion en el tejido nervioso se produce una respuesta rapida en el sitio del dano debida a la presencia de mediadores vasoactivos, incluyendo sustancia P, bradiquinina, el peptido relacionado con el gen de la calcitonina (CGRP), la IL-10 y el ON (George y col., 2004; Zochodne y col., 1999). Estos mediadores son liberados por los axones danados y ocasionan una reaccion inflamatoria e hiperemia en el microambiente. En consecuencia, se forma un denso infiltrado celular, principalmente compuesto por macrofagos, linfocitos T y mastocitos (Stoll y col., 2002; Sholtz y Woolf, 2007). Recientemente, se ha demostrado en ratones con CCN, que la infiltracion de linfocitos T esta mediada por la interleuquina IL-17A (Kleinschnitz y col., 2006). Ademas, se ha descrito en ratones knockout para esta citoquina, una reduccion significativa tanto en el numero de macrofagos en la zona de la lesion, como en los niveles de la proteina de atraccion de los macrófagos (MCP-1), lo cual se acompana de una disminucion significativa de la hiperalgesia.
Tambien se ha descrito que las citoquinas proinflamatorias pueden modular el efecto analgesico de la morfina, gracias a la activacion de las celulas gliales espinales, facilitando con ello el mantenimiento de la hiperalgesia y la alodinia (Raghavendra y col., 2002). Como evidencia de lo anterior, la aplicacion intratecal (i.t.) de minociclina, un inhibidor de la activacion microglial, atenua el dolor neuropatico inducido por lesion a nervios (Raghavendra y col., 2003).
Kleinschnitz y col. (2004) encontraron un incremento significativo del factor de necrosis tumoral (TNF-), de IL-1, IL-10 y de MCP-1, en ratones luego de efectuar la CCN. No obstante, cuando los animales reciben MK-801 por via i.p, disminuye significativamente la expresion de estas citoquinas. Por otra parte, se ha encontrado que el TNF- sensibiliza a las neuronas del aferente primario, cambiando la conductancia del canal de potasio en las neuronas del ganglio de la raiz dorsal (Czeschik y col., 2008). Ademas, se ha descrito que el tratamiento preventivo con etarnecep (un antagonista del TNF-), disminuye la hiperalgesia termica asociada a la CCN (Sommer y col., 2001).
Por otro lado, la fractalquina, una quimoquina que interactua con el receptor CX3CR1 esta involucrada en el reclutamiento microglial y en la proliferacion de astrocitos en el territorio lesionado. La administracion i.t. de fractalquina produce alodinia mecanica (Zhuang y col., 2007), mientras que la administracion de un anticuerpo neutralizante del receptor CX3CR1 retarda la aparicion de alodinia despues de la CCN (Milligan y col., 2004). Esta quimoquina media la senalizacion entre las neuronas y las glias, lo cual parece contribuir al desarrollo del dolor neuropatico (Sholtz y Woolf, 2007).
Conclusion
Alrededor del 1,5% de la poblacion mundial sufre de dolor neuropatico y a pesar de los adelantos en la fisiologia, la fisiopatologia y la farmacologia de este tipo de dolor, solo un 60% de los pacientes tratados alcanzan un manejo adecuado de esta sensacion. Son diversos los mecanismos que participan en el proceso neuropatico, agrupandose en mecanismos perifericos y centrales. La disfuncion de los canales de sodio y de potasio, el incremento en la sintesis de mediadores nociceptivos, la liberacion de glutamato, la participacion de los receptores de NMDA, la perdida de los receptores tipo opioide y la activacion de las celulas microgliales que ocurre en el asta dorsal de la medula espinal, entre otros, son mecanismos que requieren un mejor entendimiento y consideracion para la obtencion de nuevas alternativas terapeuticas. Con respecto a los modelos experimentales, sin duda han sido una contribucion de suma importancia para el conocimiento de como se genera el dolor neuropatico y el de emplear estrategias para su prevencion y tratamiento. Sin embargo, seria quimerico considerar que solo la informacion obtenida con estos modelos ayudara a solucionar las incognitas y dificultades de las distintas neuropatias. Solo la convergencia de los modelos preclinicos, los cuales proveen la informacion biologica general y los estudios clinicos, como etapa concluyente, permitira abordar los problemas observados en los pacientes y contrastar el valor clinico con los conocimientos obtenidos en los ambientes de laboratorio.

Referencias
1. Ambar M, Gratt BM. The role of nitric oxide in the physiopathology of pain. J Pain Syntom Manag 1997; 14: 225-254. [ Links ]
2. Amir R, Devor M. Functional cross-excitation in rat dorsal root ganglia. Neuroscience 2000; 95: 189-95. [ Links ]
3. Andre J, Brigitte Zeau B, Pohl M, Cesselin G, Benoliel J, Becker C. Involvement of cholecystokininergic systems in anxiety-induced hyperalgesia in male rats: Behavioral and biochemical studies. J Neuroscience 2005; 25(35):7896-7904. [ Links ]
4. Antonov S, Johnson J. Permeant ion regulation of N-methyl-Daspartate receptor channel block by Mg+2. PNAS 1999; 96(25): 14571-14526. [ Links ]
5. Attal N, Bouhassira D. Mechanism of pain in peripheral neuropathy. Acta Neurol Scand 1999; Suppl. 173: 12-24. [ Links ]
6. Baños JE, Ruiz-Barría G. La evaluación del dolor experimental en el laboratorio: los modelos de dolor neuropático en animales Rev Soc Esp Dolor 2006; 8: 542-552. [ Links ]
7. Bardoni R, Torsney C, Chi-Hun T, Prandini M, MacDermott A. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J. Neuroscienc 2004; 24(11):2774-2781. [ Links ]
8. Baron R. Mechanisms of disease: neuropathic pain--a clinical perspective. Nat Clin Pract Neurol 2006; 2(2): 95-106. [ Links ]
9. Baron R. Peripheral neuropathic pain: from mechanisms to symptoms. Clin J Pain 2000; 16(2):S12-S20. [ Links ]
10. Basbaum AI, Gautron M, Jazat F y Mayes M, Guilbaud G. The spectrum of fiber loss in a model of neuropathic pain in the rat: an electron microscopic study. Pain 1991; 47: 359-367. [ Links ]
11. Basbaum AI, Jessell T. La percepción del dolor. En: Principios de Neurociencias. Editado por: Kandel E, Schawartz J y Jessell T. Capítulo 24. Cuarta edición. McGraw-Hill. España. 2001; pp. 472-489. [ Links ]
12. Basbaum AI. Spinal mechanisms of acute and persistent pain. Reg Anesth Pain Med 2001; 24(1): 59-67. [ Links ]
13. Begon S, Pickering G, Eschalier A, Dubray C. Magnesium increase morphine analgesic effect in different experimental models of pain. Anesthesiology 2002; 96(3): 627-632. [ Links ]
14. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disordes of pain sensation like those seen in man. Pain 1988; 33: 87-107. [ Links ]
15. Bennett GJ. Animals Models of Pain. En: Methods in Pain Research. Editado por: Kruger Lawrence. Capítulo 4. CRC Press LLC. 2001; pp. 68-87. [ Links ]
16. Bennett GJ. Update on the neurophysiology of pain transmission and modulation: focus on the NMDA-receptor. J Pain Symp Manag 2000; 19(1): S2-S6. [ Links ]
17. Bereiter DA, Benetti AP. Excitatory amino acid release within spinal trigeminal nucleus after mustar oil injection into the temporomandibular joint region of the rat. Pain 1996; 67: 451-459. [ Links ]
18. Besson JM. The neurobiology of pain. Lancet 1999; 353: 1610-1615. [ Links ]
19. Bridges D, Thompson S, Rice A. Mechanism of neuropathic pain. Bri J Anaesth 2001; 87: 12-26. [ Links ]
20. Broom DC, Samad TA, Kohno T, Tegeder I, Geisslinger G, Woolf CJ. Cyclooxigenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience 2004; 124:891-900. [ Links ]
21. Calcutt NA, Chaplan SR. Spinal pharmacology of tactile allodynia in diabetic rats. Bri. J. Pharmacol 1997; (7): 1478-82. [ Links ]
22. Campbell JN, Meyer JE. Mechanisms of neuropathic pain. Neuron 2006; 52(1): 77-92. [ Links ]
23. Caraceni A, Zecca E, Bonezzi C, Arcuri E, Tur RY, Maltoni M. Gabapentin for neuropathic cancer pain: a randomized controlled trial from the gabapentin cancer pain study group. J Clin Oncol 2004; 22: 2909-917. [ Links ]
24. Chong MS, Bajwa ZH. Diagnosis and Treatment of Neuropathic Pain. J Pain Sympt Managem 2003; 25(S5): S4-S11. [ Links ]
25. Clancy J. Neurophisiology of pain. Bri J Anaesth 1995; 75: 217-227. [ Links ]
26. Coderre TJ, Katz J, Vaccarino AL, Melzack R. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 1993; 52: 259-285. [ Links ]
27. Costigan M, Woolf C. Pain: Molecular mechanisms. J Pain 2000; 1(3): 35-44. [ Links ]
28. Cruciani RA, Nieto MJ. Fisiopatología y tratamiento del dolor neuropático: avances más recientes. Rev. Soc. Esp. Dolor 2006; 13 (5): 312-327. [ Links ]
29. Czeschik JC, Hagenacker T, Schäfers M, Büsselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci Lett 2008; 434 (3): 293-8. [ Links ]
30. D'Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther 1941; 72: 74-79. [ Links ]
31. Decosterd I, Woolf C. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000; 87: 149-158. [ Links ]
32. Dickenson AH, Chapman V, Green GM. The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cord. Gen Pharmacol 1997; 28: 633-638. [ Links ]
33. Dickenson AH. Pharmacology of pain transmission and control. En: Proceedings of the 8th World Congress on Pain, Progress in Pain Research and Management . Editado por: Gebhart GF, Hammond DL, Jensen T., IASP Press 1996; 113-121. [ Links ]
34. Dingledine R, Borges K, Bowie, Traynelis S. The glutamate receptor ion channels. Pharmacol Rev 1999; 51(1): 7-61. [ Links ]
35. Dray A, Urban L, Dickenson AH. Phramacology of chronic pain. Trends Pharmacol Sci 2000; 15: 190-197. [ Links ]
36. Erichsen H, Blackburn-Munro G. Pharmacological characterisation of the spared nerve injury models of neuropathic pain. Pain 2002; 98: 151-161. [ Links ]
37. Fields HL. Pain. McGraw-Hill Book Company. New York 1987; pp 220-227. [ Links ]
38. Fischer S, Kleinschnitz C, Müller M, Kobsar I, Rollins B, Martini R. Monocyte chemoattractant protein-1 is a pathogenic component in a model for a hereditary peripheral neuropathy. Mol Cell Neurosci 2008; 37(2): 359-66. [ Links ]
39. George A, Buehl A, Sommer C. Wallerian degeneration after crush injury of rat sciatic nerve increases endo- and epineurial tumor necrosis factor-alpha protein. Neurosci Lett 2004; 372(3): 215-219. [ Links ]
40. Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs: placebo in patients with painful diabetic neuropathy. Pain 2005; 116: 109-18. [ Links ]
41. Gómez-Barrios JV. Dolor neuropático focalizado: del origen al diagnóstico. Rev Iberoamerican Dolor 2007; 2(4): 19-24. [ Links ]
42. Gwak YS, Tan HY, Nam TS, Paik KS, Hulsebosch CE, Leem JW. Activation of spinal GABA receptors attenuates chronic central neuropathic pain after spinal cord injury. J Neurotrauma 2006; 23(7): 1111-24. [ Links ]
43. Hamidi GA, Manaheji H, Janahmadi M, Noorbakhsh SM, Salami M. Co-administration of MK-801 and morphine attenuates neuropathic pain in rat. Physiol Behav 2006; 88(4-5): 628-635. [ Links ]
44. Hewett SJ, Bell SC, Hewett JA. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol Ther 2006; 112(2): 335-357. [ Links ]
45. International Association for the Study of Pain. Pain Terms: a current list with definitions and notes on usage. Pain 1994; (Supple): S215-S221. [ Links ]
46. Jagodic MM, Pathirathna S, Joksovic PM, Lee W, Nelson MT, Naik AK, Su P, Jevtovic-Todorovic V, Todorovic SM. Upregulation of the T-type calcium current in small rat sensory neurons after chronic constrictive injury of the sciatic nerve. J Neurophysiol 2008; 99(6): 3151-6. [ Links ]
47. Jander S, Pohl J, Gillen C, Stoll G. Differential expression of interleukin- 10 mRNA in Wallerian degeneration and immune-mediated inflammation of the rat peripheral nervous system. J Neurosci Res 1996; 43(2): 254-59. [ Links ]
48. Jensen MP, Chodroff MJ, Dworkin RH. The impact of neuropathic pain on health-related quality of life: review and implications. Neurology 2007; 68: 1178-82. [ Links ]
49. Joshi SK, Hernandez G, Mikusa JP, Zhu CZ, Zhong C, Salyers A, Wismer CT, Decker MW, Honore P. Comparison of antinociceptive actions of standard analgesics in attenuating capsaicin and nerve injury-induced mechanical hypersensitivity. Neuroscience 2006; 143(2): 587-96. [ Links ]
50. Kandel E, Siegelbaum SA. Integración sináptica. En: Principios de Neurociencias. Editado por: Kandel E, Schawartz J y Jessell T. Capítulo 12. Cuarta edición. McGraw-Hill. España 2001; pp 201-226. [ Links ]
51. Karlsson U, Sjodin J, Moller A, Johansson S, Wikstrom L, Nasstrom J. Glutamate-induced currents reveal three functionally distinct NMDA receptor populations in rat dorsal horn-effects of peripheral nerve lesion and inflammation. Neuroscience 2002; 112(4): 861-868. [ Links ]
52. Katz E, Gold M. Inflammatory hyperalgesia: A role for the C-fiber sensory neuron cell body?. J Pain 2006; 7(3): 170-178. [ Links ]
53. Kawamata T, Omote K. Involment of increase excitatory aminoacids and intracellular calcium concentration in the spinal dorsal horn in an animal model of neuropathic pain. Pain 1996; 68: 85-96. [ Links ]
54. Kessler W, Kirchhoff C, Reeh P, Handwerker H. Excitation of cutaneous afferent nerve endings in vitro by a combination of inflammatory mediators and conditioning effect of substance P. Exp Brain Res 1992; 91(3):467-76. [ Links ]
55. Kim KJ, Yoon TW, Chung JM. Comparison of three rodent neuropathic pain models. Exp Brain Res 1997; 113: 200-206. [ Links ]
56. Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992; 50: 355-63. [ Links ]
57. Kleinschnitz C, Brinkhoff J, Zelenka M, Sommer C, Stoll G. The extent of cytokine induction in peripheral nerve lesions depends on the mode of injury and NMDA receptor signaling. J Neuroimmunol 2004; 149: 77-83. [ Links ]
58. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol 2006; 200(2): 480-5. [ Links ]
59. Kroin JS, Buvanendran A, Watts DE, Saha C, Tuman KJ. Upregulation of cerebrospinal fluid and peripheral prostaglandin E2 in a rat postoperative pain model. Anesth Analg. 2006; 103(2): 334-43. [ Links ]
60. Lee BH, Won R, Baik EJ, Lee SH, Moon CH. An animal model of neuropathic pain employing injury to the sciatic nerve branches. Neuroreport 2000; 11: 657-661. [ Links ]
61. Lee CJ, Bardoni R, Tong CK, Engelman HS, Josefh DJ, Magherini PC, MacDermott AB. Functional expression of AMPA receptors on central terminals of rat dorsal root ganglion neurons and presynaptic inhibition of glutamase release. Neuron 2002; 35: 135-146. [ Links ]
62. Lenz FA, Gracely RH, Baker FH, Richarson RT, Dougherty PM. Reorganization of sensory modalities evoked by microstimulation in region of the thalamic principal sensory nucleus in patients with pain due to nervous system injury. J Comprar Neurol 1998; 399: 125-138. [ Links ]
63. Li Y, Dorsi MG, Meyer MA, Belzberg AJ. Mechanical hiperalgesia after L5 spinal nerve lesion in the rat is not depending on input from injured nerve fibres. Pain 2000; 85: 493-502. [ Links ]
64. Liu CN, Devor M, Waxman SG, Kocsis JD. Subthreshold oscillations induced by spinal nerve injury in dissociated and cutaneous afferents of mouse DRG. J Neurophysiol 2002; 87: 2009-2017. [ Links ]
65. Liu H, Mantyh PW, Basbaum AI. NMDA receptors regulation of substance P release from primary afferent nociceptores. Nature 1997; 386: 721-724. [ Links ]
66. Ma W, Eisenach JC. Intraplantar injection of a cyclooxygenase inhibitor ketorolac reduces immunoreactivities of substance P, calcitonin gene-related peptide, and dynorphin in the dorsal horn of rats with nerve injury or inflammation. Neuroscience 2003; 121(3): 681-90. [ Links ]
67. Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Neuroscience 2005; 6: 521-532. [ Links ]
68. Mayer DJ, Mao J, Holt J, Price DD. Cellular mechanisms of neuropathic pain, morphine tolerance, and their interactions. Proc Natl Acad Sci 96 1999; 7731-7736. [ Links ]
69. McHugh JM, McHugh WB. Pain: neuroanatomy, chemical mediators, and clinical implications. AACN Clin Issues 2002; 11(2): 168-78. [ Links ]
70. Merskey H, Bogduk N. Classification of chronic pain. IASP Press, Second Ed., Seattle 1994; pp 209-214. [ Links ]
71. Mielke S, Sparreboom A, Steinberg SM, Gelderblom H, Unger C, Behringer D, Mross K. Association of Paclitaxel pharmacokinetics with thedevelopment of peripheral neuropathy in patientswith advanced cancer. Clin Cancer Res 2005; 11(13): 4843-4850. [ Links ]
72. Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, OConnor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci 2004; 20: 2294-2302. [ Links ]
73. Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev 2006; 51(2): 240-64. [ Links ]
74. Morisett V, Nagy F. Ionic basis for plateau potentials in deep dorsal horn neurons of the rat spinal cord. J Neurosci 1999; 19(17): 7309-7316. [ Links ]
75. Naik AK, Tandan SK, Kumar D, Dudhgaonkar SP. Nitric oxide and its modulators in chronic constriction injury-induced neuropathic pain in rats. Eur J Pharmacol 2006; 530(1-2): 59-69. [ Links ]
76. Nakamura S, Atsuta Y. Electrophysiological study on primary afferent properties of a chronic constriction nerve injury model in spinal rats. J Orthop Sci 2004; 9(4): 386-91. [ Links ]
77. Nakatsuka T, Chen M, Takeda D, King C, Ling J, Xing H, Ataka T, Vierck C, Yezierski R, Gu JG. Substance P-driven feed-forward inhibitory activity in the mammalian spinal cord. Mol Pain 2005; 1: 20-29. [ Links ]
78. Narita M, Miyoshi K, Narita M, Suzuki T. Changes in function of NMDA receptor NR2B subunit in spinal cord of rats with neuropathy following chronic ethanol consumption. Life Sci 2007; 80(9): 852-859. [ Links ]
79. Omana-Zapata I, Khabbaz MA, Hunter JC, Clarke DE, Bley KR. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 1997; 72: 41-49. [ Links ]
80. Parsons CG. NMDA receptors as targets for drug action in neuropathic pain. Eur J Pharmacol 2001; 429: 71-78. [ Links ]
81. Polgar E, Gray S, Riddell JS, Todd AJ. Lack of evidence for significant neuronal loss in laminae I-III of the spinal dorsal horn of the rat in the chronic constriction injury model. Pain 2004; 111(1-2): 144-50. [ Links ]
82. Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 2001; 94: 293-304. [ Links ]
83. Qian A, Antonov S, Johnson J. Modulation by permeant ions of Mg+2 inhibition of NMDA activated whole-cell currents in rat cortical neurons. J Physiol 2002; 538(1): 65-77. [ Links ]
84. Qian A, Johnson J. Channel gating of NMDA receptors. Physiol Behav 2002; 577-582. [ Links ] 85. Raghavendra V, Rutkowski, DeLeo J. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neuros 2002; 22(22): 9980-9989. [ Links ]
86. Raghavendra V, Tanga F, Rutkowski MD, and DeLeo, JA. Anti-hyperalgesic and morphine-sparing actions of propentofylline following peripheral nerve injury in rats: mechanistic implications of spinal glia and proinflammatory cytokines. Pain 2003; 104: 655-664. [ Links ]
87. Rajagopal A, Vassilopoulou-Sellin R, Palmer JL, Kaur G, Bruera E. Symptomatic hypogonadism in male survivors of cancer with chronic exposure to morphine. Cancer 2004; 100: 851-8. [ Links ]
88. Rexed B. The cytoarchitectonic organization of the spinal cord in the cat. J Comp Neurol 1954; 96:415-495. [ Links ]
89. Ricote M, Valledor AF, Glass CK. Decoding transcrip¬tional programs regulated by PPARs and LXRs in the macrophages: Effects on lipid homeostasis, inflamma¬tion, and atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 230-39. [ Links ]
90. Ruppersberg J, Kitzing E, Schoepfer R. The mechanism of magnesium block of NMDA receptors. Semin Neurosc 1994; 6(2): 87-96. [ Links ]
91. Sabadowski R, Galvez R, Cherry DA, Jacquot F, Vincent E, Maisonope P. Pregabalin reduces pain and improves mood disturbances in patients with post herpetic neuralgia, results of a randomised placebo- controlled clinical trial. Pain 2004; 109: 26-35. [ Links ]
92. Sanoja R, Vanegas H, Tortorici V. Critical role of RVM in the early spinal events leading to CCI neuropathy in rats. J Pain 2008; 9(6): 532-542. [ Links ]
93. Schafer M, Zhou L, Stein C. Cholecystokinin inhibits peripheral opioid analgesia in inflamed tissue. Neuroscience 1998; 82(2): 603-611. [ Links ]
94. Scholtz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007; 10(11): 1361-8. [ Links ]
95. Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990; 43: 205-211. [ Links ]
96. Shields SD, Eckert WA, Basbaum AI. Spared Nerve Injury Model of Neuropathic Pain in the Mouse: A Behavioral and Anatomic Analysis. J Pain 2003; 4(8): 465-470. [ Links ]
97. Siau C, Xiao W, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: Loss of epidermal innervation and activation of Langerhans cells. Exp Neurol 2006; 201(2): 507-14. [ Links ]
98. Skyba DA, Lisi TL, Sluka KA. Excitatory amino acid concentrations increase in the spinal cord dorsal horn after repeated intramuscular injection of acidic saline. Pain 2005; 119: 142-149. [ Links ]
99. Smith DG, Ehde DM, Hanley MA, Campbell KM, Jensen MP, Hoffman AJ, et al. Efficacy of gabapentin in treating chronic phantom limb and residual limb pain. J Rehabil Res Dev 2005; 42: 645-54. [ Links ]
100. Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV. Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res 2001; 913: 86-89. [ Links ]
101. Staats PS, Yearwood T, Charapata SG, Presley RW, Wallace MS, Byas-Smith M. Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS: a randomized controlled trial. JAMA 2004; 291(1): 63-70. [ Links ]
102. Stoll G, Jander S, Myers RR. Degeneration and regeneration of the peripheral nervous system: from Augustus Wallers observations to neuroinflammation. J Peripher Nerv Syst 2002; 7: 13-27. [ Links ]
103. Streit, W.J. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002; 40: 133-139. [ Links ]
104. Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transsection, and strychnine. Pain 1990; 42: 205-213. [ Links ]
105. Sun, Q, Tu, H, Xing, G, Hans, J y Wan Y. Ectopic discharges from injured nerve fibers are highly correlated with tactile allodynia only in early, but not late, stage in rats with spinal nerve ligation. Exp Neurol 2005; 191(1): 128-36. [ Links ]
106. Tandrup T, Woolf CJ, Coggeshall RE. Delayed loss of small dorsal root ganglion cells after transsection of the rat sciatic nerve. J Comp Neurol 2000; 422: 172-180. [ Links ]
107. Tassorelli C, Greco R, Wang D, Sandrini G, Nappi G. Prostaglandins, glutamate and nitric oxide synthase mediate nitroglycerin-induced hyperalgesia in the formalin test. Eur J Pharmacol 2006; 534(1-3): 103-107. [ Links ]
108. Thomson LM, Zeng J, Terma GW. An N-methyl-D-aspartate receptor mediated large, low-frequency, spontaneous excitatory postsynaptic current in neonatal rat spinal dorsal horn neurons. Neuroscience 2006; 141(3): 1489-501. [ Links ]
109. Tomiyama M, Furusawa K, Kamijo M, Kimura T, Matsunaga M, Baba M. Upregulation of mRNAs coding for AMPA and NMDA receptor subunits and metabotropic glutamate receptors in the dorsal horn of the spinal cord in a rat model of diabetes mellitus. Brain Res Mol Brain Res 2005; 136(1-2): 275-81. [ Links ]
110. Tortorici V. Fisiopatología del dolor neuropático. En; Consenso Venezolano de Dolor Neuropático. Asociación Venezolana para el Estudio del Dolor (AVED). Caracas 2008; pp. 11-28. [ Links ]
111. Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, Hansson P, Hughes R, Nurmikko T, Serra J. Neuropathic pain. Redefinition and a grading system for clinical and research purposes. Neurology 2008; 70(18): 1630-1635. [ Links ]
112. Ultenius C, Linderoth B, Mcyerson B, Wallin J. Spinal NMDA receptor phosphorylation correlates with the presence of neuropathic signs peripheral nerve injury in the rat. Neuroscienc Lett 2006; 222: 85-70. [ Links ]
113. Vásquez E, Bar K J, Ebersberger A, Klein B, Vanegas H, Schaible HG. Spinal prostaglandins are involved in the development but not the maintenance of inflammation-induced spinal hyperexcitability. J Neurosci 2001; 21: 9001-9008. [ Links ]
114. Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci U.S.A. 1999; 96 (14): 7635-7639. [ Links ]
115. Weber WEJ. Randomised controlled trial of gabapentin in Complex Regional pain Syndrome type I. BMC Neurol 2004; 4(13): doi:10.1186/1471-2377-4-13. [ Links ]
116. White DM, Cousins MJ. Effect of subcutaneous administration of calcium channel blockers on nerve injury-induced hyperalgesia. Brain Res. 1998; 801(1-2): 50-8. [ Links ]
117. Wilson RC. The use of low-dose trazodone in the treatment of painful diabetic neuropathy. Am Podiatr Med Assoc 1999; 89(9): 468-71. [ Links ]
118. Woolf CJ, Ma Q. Nociceptors--noxious stimulus detectors. Neuron 2007; 55(3): 353-64. [ Links ]
119. Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symtoms, mechanisms, and management. Lancet 1999; 353: 1959-964. [ Links ]
120. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science 2000; 288: 1765-1768. [ Links ]
121. Woolf CJ, Shortland P, Reynolds ML. Central regenerative sprouting: the reorganization of the central terminals of myelinated afferents in the rat dorsal horn following peripheral nerve section or crush. J Comp Neurol 1995; 360: 121-134. [ Links ]
122. Woolf CJ, Thompson SW. The induction and maintenance of central sensibilization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 1991; 44: 293-299. [ Links ]
123. Woolf CJ, Wall PD. Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci 1986; 6(5): 1433-42. [ Links ]
124. Woolf CJ. Dissecting out mechanisms responsible for peripheral neuropathic pain: Implications for diagnosis and therapy. Life Sci 2004; 74:2605-2610. [ Links ]
125. Woolf CS, Chong MS. Preemptive analgesia-treating postoperative pain by preventing the establishment of central sensitization. Anesth Analg 1993; 77(2): 362-79. [ Links ]
126. Wright A. Recent concepts in the neurophysiology of pain. Man Ther 1999; 4: 196-202. [ Links ]
127. Wu HE, Schwansinger ET, Hong JS, Tseng LF. Pretreatment with antiserum against dynorphin, substance P, or cholecystokinin enhances the morphine-produced anti-allodynia in the sciatic nerve ligated mice. Neurosci Lett 2005; 386(1): 46-51. [ Links ]
128. Xiao WH, Bennett GJ. Magnesium suppresses neuropathic pain responses in rats via a spinal site of action. Brain Res 1994; 666(2): 168-172. [ Links ]
129. Xu XJ, Alster P, Wu WP, Hao JX, Wiesenfeld-Hallin Z. Increased level of cholecystokinin in cerebrospinal fluid is associated with chronic pain-like behavior in spinally injured rats. Peptides 2001; 22(8): 1305-1308. [ Links ]
130. Yaksh TL. Spinal systems and pain processing: development of novel analgesic drugs with mechanistically defined models. Trends Pharmacol Sci 1999; 329-337. [ Links ]
131. Zakrzewska JM, Patsalos PN. Long-term cohort study comparing medical (oxcarbazepine) and surgical management of intractable trigeminal neuralgia. Pain 2002; 95(3): 259-66. [ Links ]
132. Zernikow B, Smale H, Michel E, Hasan C, Jorch N, Andler W. Pediatric cancer pain management using the WHO analgesic ladder results of prospective analysis from 2265 treatment days during a quality improvement study. Eur J Pharmacol 2006; 10: 587-595. [ Links ]
133. Zhao Z, Chen SR, Eisenach JC, Busija DW, Pan HL. Spinal cyclooxygenase- 2 is involved in development of allodynia after nerve injury in rats. Neuroscience 2000; 97(4): 743-8. [ Links ]
134. Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun 2007; 21: 642-651. [ Links ]
135. Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol 2001; 429: 23-37. [ Links ]
136. Zochodne, DW, Levy D, Zwiers H, Sun H, Rubin I, Cheng C, Lauritzen M. Evidence for nitric oxide and nitric oxide synthase activity in proximal stumps of transected peripheral nerves. Neuroscience 1999; 91: 1515-1527. [ Links ]
137. Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res 2005; 1034(1- 2): 11-24. [ Links ]
