










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Facultad de Medicina
versión impresa ISSN 0798-0469
RFM v.23 n.2 Caracas jul. 2000
ASIMETRÍA EN EL HIPERPARATIROIDISMO SECUNDARIO RENAL. UNA REVISIÓN TEÓRICO ANALÍTICA
L Fernández1, A Blanco2, M Bustamante2 y J Benchimol3.
RESUMEN
La asimetría del hiperparatiroidismo en la insuficiencia renal crónica es muy infrecuente y está poco estudiado, algunos autores lo atribuyen a inactivación del gen supresor tumoral del cromosoma 11, activación de oncogénes, modificación en los receptores de vitamina D y otros factores tróficos.
A propósito de una revisión teórico - analítica de los archivos de Anatomía Patológica del Hospital Miguel Pérez Carreño(HMPC), Caracas - Venezuela período 1994-1998. Se encontró de 21000 biopsias generales, dos casos de esta patología.
Se realizó una revisión teórico-analítica para destacar los planteamientos hechos a la fecha sobre dicho tópico.
Palabras Claves: Hiperparatiroidismo, Insuficiencia renal crónica, Asimetría.
ABSTRACT
The hyperparathyroidism asymmetry in the chronic kidney failure is not very frequent and few studies have been reported in this subject. Some authors attribute this to the inactivation of the tumoral supressor gene from the chromosome 11, activation of oncogenes, modification of calcium receptors and other "trofic" factors.
A trial of archives of the service of pathology of the Miguel Pérez Carreño hospital, Caracas-Venezuela, during the years, 1994 to 1998, was made and two cases of this pathology were found among the 21000 general biopsias reviewed.
A theoretic-analytic review is made to emphasize the actual expositions in this matter.
Key Words: Hyperparathyroidism, Chronic kidney failure, Asymmetry.
__________________
IntroducciÓn
El hiperparatiroidismo renal está ampliamente descrito en la literatura, sin embargo, la hiperplasia difusa nodular autónoma asimétrica que se ve en las paratiroides es poco conocida, involucrándose a factores como pérdida alélica de cromosoma 11(1), activación de oncogenes(2,3), factores tróficos (4,5,6,7,8) y modificación de receptores de vitamina D en su génesis(9).
A propósito de una revisión en el archivo del Hospital Miguel Pérez Carreño, Caracas-Venezuela, período 1994-1998, de un universo de 21000 biopsias generales se encontraron dos casos de hiperparatiroidismo secundario renal asimétrico. Esto motiva a realizar una revisión teórico-analítico para destacar los planteamientos hechos a la fecha para explicar esta patología.
Materiales y mÉtodos
Se revisó el archivo de anatomía patológica del Hospital Miguel Pérez Carreño, Caracas-Venezuela, período 1994-1998. En un universo de 21000 biopsias generales se encontraron dos casos de hiperparatiroidismo secundario urémico asimétrico en las 4 paratiroides de cada caso estudiado. Se trata de dos pacientes, uno masculino y uno femenino de 22 y 25 años de edad respectivamente, con insuficiencia renal crónica de larga data, 9 y 10 años, en los que se intento tratamiento con etanol local no siendo efectivo por lo que se procedió a la paratiroidectomía, implantándose fragmentos de la paratiroides más pequeña en antebrazo, siendo positivo los niveles medidos de la parathormona un año y medio después de dicha intervención. Esperan transplante renal actualmente.
Estudio de anatomÍa patolÓgica
Descripción macroscópica
En los dos pacientes se encontraron cuatro glándulas parotidoides, cuyo tamaño variaba entre 2,6*1,8*0,7 cm y 1*1cm. Son de color pardo amarillento, al corte se observaron focos nodulares y hemorrágicos, firmes (Fig. 1).
Fig. 1. Se destaca la asimetría de las cuatro paratiroides de uno de los pacientes.

Descripción microscópica
Los cortes histológicos muestran diferentes patrones, difuso y nodular. Se evidenciaron cambios marcados en el tamaño de los núcleos con citoplasma abundante granular y marcada eosinofília (Adenoma oxífilico). (Fig. 2 y 3). Igualmente se apreciaron cambios nucleares marcados en las célular principales de citoplasma claro, disposición uniforme perivascular (hiperplasia de células principales).(Fig. 4). Estos cambios fueron similares en los dos pacientes.
Fig. 2. Células oxifilicas con marcada macrocariosis, disposición cromatínica irregular. HE A: 40* 10.
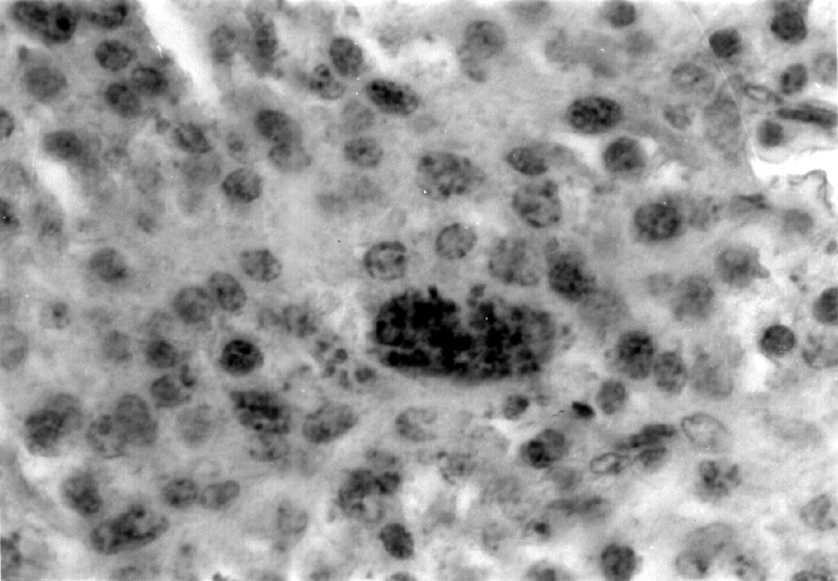
Fig. 3. Panorámica de paratiroides. Patrones diferentes , nodular y difuso, conformado por células oxifílicas (Adenoma) HE. 10* 10.

Fig. 4. Células principales, macrocariosis marcada, citoplasma claro, disposición perivascular (Hiperplasia Células principales). HE. A: 25* 10.

DiscusIÓn
Las paratiroides son las principales reguladoras de la concentración del calcio plasmático y del flujo de calcio en el cuerpo(10). Son generalmente 4 aunque un 10% de las personas tienen sólo 2 ó 3(11). Se originan de la tercera a cuarta bolsa faríngea comenzando su actividad reguladora en la quinta semana de gestación(12). Histológicamente tienen dos tipos de células, principales y oxífilas, siendo las primeras las encargadas de la producción y secreción de la parathormona (HPT); de 5 a 7 años después del nacimiento las células oxífilas se diferencian, siendo su función más que producir HPT, almacenar energía a través de gránulos de glucógeno(10).
Las funciones de dicha hormona son: acelerar la remoción de calcio del hueso, aumentar la absorción de calcio y fósforo del tracto gastrointestinal, aumentar la reabsorción de calcio desde el túbulo distal del riñón, inhibir la reabsorción de fosfato en el túbulo proximal y estimular la síntesis en los túbulos renales de metabolitos de la vitamina D que poseen acción de reabsorber hueso(10).
La secreción, síntesis y degradación de la HPT dentro de las glándulas es regulada por el magnesio, histamina, adrenalina, metabolitos de la vitamina D, fósforo y por el calcio(10). La secreción y degradación intracelular de HPT tiene diferente sensibilidad de ser inhibida por el calcio, es así como, con valores de calcio alto la secreción de fragmentos de HPT se inhibe menos que la hormona intacta(14,15). El fósforo posee un efecto directo al aumentar la síntesis y secreción de esta hormona(13) in vivo e in vitro independientemente de los niveles de calcio ionizado y 1,25 D3(5).
Existen patología que afectan esta glándula, como es el caso de la insuficiencia renal (hiperparatiroidismo urémico), la deficiencia de vitamina D y la osteomalacia que conllevan al hiperparatiroidismo secundario(11,16) y producen gran variación en la talla de esta glándula(17), como se vió en los dos casos estudiados.
El origen del hiperparatiroidismo secundario en la insuficiencia renal se atribuye a:
a) Retención de fósforo(5,18).
b) Hipocalcemia, sobre la cual todavía no hay datos indicativos de que grado, duración o frecuencia de ella es necesaria para producir hiperplasia paratiroidea en humanos(19).
c) Oncogénes.
d) Genes supresores.
e) Factores tróficos como:
• Contenido de receptores de vitamina D, que dependen del patrón histológico más que del peso grandular(4) y las hacen resistentes a la acción de 1,25 D3 en sangre, para suprimir los niveles de hormona paratiroidea en la insuficiencia renal avanzada(5).
• Expresión del factor de crecimiento tumoral alfa y del receptor del factor de crecimiento epidermal, que promueven la proliferación de células paratiroideas, tal vez por un mecanismo autocrino. Estos factores se encuentran tanto en glándulas paratiroideas hiperplásicas como adenomatosas(8).
Todos lo anterior conduce a una hiperfunción paratiroidea compensadora con hiperplasia, compuesta básicamente por células principales mezcladas con áreas de células oxífilas las cuales también sintetizan y secretan HPT y pueden presentar defectos en la cadena respiratoria que favorece mutaciones del DNAm, posible causa de transformación de esta célula, lo cual contribuye a la patofisiología del hiperparatiroidismo urémico (20). En dicha entidad se observa una progresión del tejido glandular normal a hiperplasia difusa y policlonal, luego o nodular y monoclonal donde en cada nódulo(21) existe disminución de la expresión del RNAm del receptor del calcio que pudiera ser compatible con una menor síntesis y secreción de HPT por calcio plasmático que en el tejido paratiroideo normal(9). Estos nódulos además pueden contener un tipo celular único con potencial proliferativo agresivo(22) hasta llegar al adenoma(3). Lo anterior expuesto puede ser consecuencia de una pérdida alélica del cromosoma 11, similar a lo encontrado en algunos adenomas esporádicos(1) o en tumores paratiroideos que forman parte de un síndrome familiar tal como la neoplasia endocrina múltiple familiar tipo I (MEN I)(23).
Los cambios morfológicos en el hiperparatiroidismo secundario imitan a los de la hiperplasia primaria. Habitualmente todas las glándulas están afectadas aunque de forma infrecuente una, dos o incluso tres pueden estar conservadas. En nuestro caso una permaneció inalterable y tres se modificaron en morfología, macro y microscópicamente con las variantes morfológica difusa y nodular. Esta transición histológica ha sido descrita previamente en la literatura.
Está descrito que si se revierte la condición que originó la hiperplasia, las glándulas vuelven a la normalidad, excepción a este hecho son las hiperplasias secundarias de larga duración que implican cambios nodulares autónomos(1) a través del cual pudiera explicarse porque pacientes con hiperplasia nodular en hiperparatiroidismo renal son refractarios al tratamiento médico requiriendo paratiroidectomía(22) para conseguir el control endocrino de la hipercalcemia tal como ocurrió en la historia clínica de los pacientes estudiados, esto se conoce como hiperparatiroidismo terciario(1,11,16,22). En resumen esta entidad clínica es posiblemente debida a la formación de un adenoma en una de las glándulas hiperplásicas(1). Otros estudios ofrecen nuevas evidencias de que la proliferación paratiroidea autónoma en pacientes urémicos puede desarrollar sobrecrecimiento por un tumor monoclonal(1).
Lo aconsejable sería prevenir estos cambios y la transición previamente descrita mediante la restricción de fósforo en la dieta ya que este independientemente de niveles séricos de calcio ionizado y 1,25 (OH2D3) tiene un efecto sobre la función paratiroidea(18,24). Además, se ha visto que la dieta rica en fosfato aumenta la talla y número de células paratiroideas en ratas adultas aunque la contribución relativa a la hipertrofia e hiperplasia es desconocida(13).
Otro método sería dar pulsos de alfacalcidol oral tempranamente en el curso de la insuficiencia renal puesto que la respuesta de inhibición de la hiperplasia paratiroidea autónoma es buena(25), sin embargo, en glándulas paratiroideas grandes con hiperplasia nodular se ha visto que son más resistentes a la terapia de pulsos de calcitriol que las pequeñas glándulas, esa respuesta a la terapia de calcitriol es restaurada a través de la destrucción selectiva de glándulas paratiroideas grandes (mayor de 0,5 cm) por la inyección de etanol a través de guía ultrasonográfica, además, inyecciones directas de solución de calcitriol dentro de grandes glándulas eran también efectivas en suprimir HPT y restaurar respuestas a calcitriol(7). En los casos estudiados se intentó con etanol siendo infructuoso y se procedió a la remoción quirúrgica implantándose en el antebrazo fragmentos de la glándula más pequeña que era normal macroscópica y microscópicamente.
Algunos autores han evaluado la incidencia de la recurrencia del hiperparatiroidismo en relación al rasgo histopatológico; análisis de DNA han indicado que la hiperplasia paratiroidea nodular tiene mayor potencial de crecimiento que la difusa, es por ello que para prevenir la recurrencia el tipo nodular de hiperplasia no debe ser autotransplantado(27). También debe tomarse en cuenta que las glándulas paratiroideas grandes están compuesta de hiperplasia nodular con proliferación de células monoclonales(28).
En este trabajo se quiere destacar la asimetría como respuesta a un mismo estímulo que causa progresión de tejido normal a hiperplasia difusa y de esta a hiperplasia nodular y adenoma en la insuficiencia renal, estos cambios fueron descritos por Tominanga(26) en el hiperparatiroidismo primario no familiar (más frecuentemente visto en ancianos).
Llama a la reflexión como ante estímulos iguales, repetitivos y de larga data se observa que hay tejido glandular que permanece inalterable en tamaño y otros se modifican tanto en tamaño como en patrones microscópicos diferentes.
Agradecimientos
Agredecemos la colaboración invaluable del Sr. Luis Aguilar, Sra. Virginia Guau y Dra. Judith Barroso.
REFERENCIAS BibliográfICaS
1. Falcheth A, et al. Progression of uremic Hyperparathyroidism involves Allelic loss on chromosome 11. Journal of clinical Endocrinology and Metabolism. 1993; 76 (1): 139-144. [ Links ]
2. Brandi ML. Molecular mechanism of parathyroid hyperplasia and neoplasia. Horm Res.1997; 47 (6):194-8. [ Links ]
3. Tominaga Y, Takagi-H: Molecular genetics of hyperparathyroid disease. Curr- Opin-Nephrol-Hypertens. 1996;5 (4):336-41. [ Links ]
4. Chou H, et al: Case of a parathyroidectomized patient observed longitudinally by ultrasonography, relationship between the growth rates and 1,25 dihydroxyvitamin D3 receptor contents in the parathiroid glands. Nephron.1996;73 (4):695-700. [ Links ]
5. Slatopolsky EY, Delmez JA: Pathogenesis of seconday hyperparathyroidism. Nephrol Dial Trasplant. 1996; 11(3): 130-135. [ Links ]
6. Fukagawa M, et al. Molecular basis for the management of secondary hyperparathyroidism in chronic renal failure. Artif organs; 1995; 19 (12): 1210-4. [ Links ]
7. Fukagawa and Kurokawa K: Pathogenesis and medical treatment of secondary hyperparathyroidism. Semin Surg Oncol. 1997; 13(2): 73-7. [ Links ]
8. Gogusev J, Duchambon P, et al. De novo expression of trasforming Growth factor-alpha in parathyroid gland tissue of patients with primary or secundaryuraemic hyper-parathyroidism. Nephrol Dial Transplant. 1996; 11 (11): 2155-62. [ Links ]
9. Gogusev J, et al. Depressed expression of calcium receptor in parathyroid gland tissue of patients with hyperparathyroidism. Kidney Int. 1997; 51(1): 328-36. [ Links ]
10. Berne Levi: Fisiología. Editorial médica Panamericana 2da. edición. Buenos Aires. 1987; 956-976. [ Links ]
11. Robbins: Patología estructural y funcional. Interamericana Mc Graw - Hill, 5ta. edición, España. 1995;1257-1262. [ Links ]
12. Keith Moore: Embriología clínica. Interamericana, 4ta. edición, México. 1988; 197. [ Links ]
13. Wang Q, et al. Phosphate administration increases both size and number of parathyroid cells in adult rats. Calcif tissue Int. 1996; 58 (1): 40-4. [ Links ]
14. D’Amour-P, et al. The modulation of circulating parathyroid hormone inmunoheterogeneity in man by ionized calcium concentration. J-Clin-Endocrinol-Metab. 1992; 20: 101-115. [ Links ]
15. Mallette - LE, et al. Malignancy hypercalcemia: evaluation of parathyroid function and response to treatment. Am - J - Med - Sci. 1991; 302 (4): 205-10. [ Links ]
16. Rubin/Forber: Patología. Editorial Panamericana. México. 1ra. edición. 1990; 1032-1033. [ Links ]
17. Salusky IB, Goodman WG. In vivo studies of parathyroid gland function in secondary hyperparathyroidism. Adv Nephrol Necker Hosp; 1996; 25: 289-302. [ Links ]
18. Denda M, Finch J y Slatopolsky E: Phosphorus accelerates the development of parathyroid hyperplasia and secondary hyperparathyroidism in rats with renal failure. Am J. Kidney Dis. 1996; 28(4): 586-602. [ Links ]
19. Mallette Le, Hollis-BW, et al. Ten, weeks of intermittent hypocalcemic stimulation does not produce functional parathyroid hyperplasia. Am J Med. Sci. 1991; 302(3): 138-41. [ Links ]
20. Tanaka Y, Funahasshi H, et al. Oxyphil cell function in secondary parathyroid hyperplasia. Nephron. 1996; 73(4): 580-6. [ Links ]
21. Müller-Höcker, et al. Defects of respiratory chain in oxyphil and Chief cells of the normal parathyroid and in hyperfunction. Hum Pathol. 1996; 27(6). 532-4. [ Links ]
22. Tominanga Y, Kohara S, et al. Clonal analysis of nodular parathyroid hyperplasia in renal hyperparathyroidism. World J. Surg. 1996; 20(7): 744-50. [ Links ]
23. Friedman, et al. Allelic loss from chromosome 11 in parathyroid tomors. Cancer Res. 1992; 52: 6804. [ Links ]
24. Combe C, et al. Role of phosphorus in hyperparathyroidism secondary to non-dialysed adult chonic renal insufficiency. Nephrologie; 1996;17(3): 149-56. [ Links ]
25. Koskimies O and Ala - Houhala M: Alphacalcidol oral pulses are effetive in secondary hyperparathyroidism prior to dialysis. Clin Nephrol. 1996; 46 (1): 70-1. [ Links ]
26. Tominaga-Y, et al. Histological and clinical features of non - familial primary parathyroidism hyperplasia. Pathol Res pract. 1992; 188 (1-2): 115-22. [ Links ]
27. Tominaga-Y, et al: Recurrent renal hyperparathyroidism of autogrfted parathyroid tissue. World J Surg. 1992; 16(4): 595-602. [ Links ]
28. Fukagawa M, et al: Ultrasonographic intervention of parathyroid hyperplasia in chronic dialysis patients: a theoretical approach. Nephrol Dial Transplant; 1996;11 (suppl 3): 125-9. [ Links ]
