










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Oncología
versión impresa ISSN 0798-0582
Rev. venez. oncol. v.18 n.4 Caracas dic. 2006
TUMOR NEUROECTODÉRMICO PRIMITIVO DE MAXILAR: PRESENTACIÓN DE UN CASO
Lourdes Peña Sevilla1, Lilibeth Piñate1, María Granados1, Alfonso López 1, Claudia Sánchez1, Elien Morao2, Reina Arteaga1, Nilda Rojas1, Ruth Gómez1
1SERVICIO DE ONCOLOGÍA PEDIÁTRICA, INSTITUTO ONCOLÓGICO DR. LUIS RAZETTI, 2HOSPITAL MILITAR DR. CARLOS ARVELO, CARACAS, VENEZUELA
Correspondencia: Dra. Lourdes Peña Sevilla Servicio de Pediatría, Instituto Oncológico Dr. Luis Razetti, Calle Real de San José del Ávila Cotiza, Caracas, Venezuela Teléfono: (212) 520810
RESUMEN
El tumor neuroectodérmico primitivo periférico forma parte de la familia de tumores sarcoma de Ewing representando el 5 % de los mismos, en maxilar superior es infrecuente. Se reporta este caso clínico de un adolescente masculino, de 15 años referido a nuestro centro con pérdida de peso, tumoración en cuadrante superior izquierdo de cavidad bucal, de 6 cm de diámetro, que ocupa el 50 % de la misma, sobre infectada, con áreas de necrosis. La biopsia reportó: tumor de células redondas indiferenciadas. Se observó rápido crecimiento tumoral que triplica su tamaño en 8 días, protruyendo por cavidad oral, la ocupa en totalidad, tendiente a obstrucción de la vía aérea; ameritó traqueotomía y gastrostomía de emergencia. Los estudios de inmunohistoquímica concluyen: tumor neuroectodérmico primitivo (inmunorreactivo a CD99, enolasa neuroespecífica y cromogranina), recibe dos ciclos de ciclofosfamida, vincristina, actinomicina D, ifosfamida y etoposido, con progresión de la enfermedad y deterioro del estado físico, por lo que recibe radioterapia 6 000 cGy con cisplatino, lográndose condiciones favorables a la cirugía: Maxilectomía bilateral con preservación del piso de la órbita. El estudio anatomopatológico reportó tejido fibromixoide con abundante vascularización sin evidencia de malignidad, márgenes libres. Actualmente, 7 meses sin evidencia de enfermedad en espera de prótesis palatina para la restitución de deglución funcional. En localizaciones infrecuentes debe el tratamiento ser multidisciplinario e individualizado, incluyendo las tres modalidades terapéuticas (quimioterapia, radioterapia y cirugía).
PALABRAS CLAVE: Cáncer, tumor neuroectodérmico primitivo, cavidad oral, maxilar, tratamiento, cirugía, radioterapia, quimioterapia.
SUMMARY
Periferical neroectodermic primary tumor is a part of the sarcoma Ewing family and represents 5 % of them. The superior maxillary location is not frequent, we report these clinical case of a male teenage of 15 years old sent to our hospital with loss weigh, tumor in the oral cavity in the left superior quadrant of the oral cavity, with a diameter of 6 cm, that it occupies 50 % of the same one with sobreinfection, with necrosis. The biopsy reported: tumor of undifferentiated round cells. We observed a growth of the tumor that triples its size in 8 days, it occupies it in totality, with tendency to obstruction of the air way, needed emergency traqueostomy and gastrostomy. The inmunohistoquimical studies reveal primary neuroectodermic tumor (inmunoreactive to CD99 enolasa neurospecific and cromagine), received two cycles of ciclofosfamide, vincristine, actinomicyn D, ifosfamida and etoposid, with disease progression and depletory of his physical status; receive radiation therapy 6 000 cGy with platinum, obtaining favorable condition to surgery: A bilateral maxilectomy with preservation of the floor of the orbit. The anatomopathological study report fibromixoide weave a lot of vascularization without evidence of malignity, free margins. Actually, 7 months without evidence of disease waiting for platinum prosthesis to restituted the functional deglution. In the inusual localizations, the treatment has to be multidisciplinary, individualized, and include the three treatment modalities (Quemotherapy, radiation therapy and surgery).
KEY WORDS: Cancer, primary neuroectodermic tumor, oral cavity, maxillary, treatment, surgery, quemotherapy, radiation therapy.
Recibido: 30/07/2005 Revisado: 18/11/2005 Aceptado para publicación: 12/03/2006
INTRODUCCIÓN
El sarcoma de Ewing fue descrito por James Ewing en 1921(1), quien caracterizó este tumor dentro del árbol de los tumores óseos y le otorgó un origen epitelial (vascular óseo), por lo cual, lo denominó endotelioma de hueso, destacándose por su radiosensibilidad y posterior recidiva(2). En 1928, Charles Oberling promulgó que el sarcoma de Ewing, derivaba de una célula mesenquimal pluripotencial de la médula ósea. El origen del tumor fue motivo de controversia durante los años 1930 y 1940 hasta que, en 1957, Fritz Schajowicz publicó en Argentina una serie de reportes donde muestra que los sarcomas de Ewing tienen un alto grado de indiferenciación y abundante contenido de glicógeno citoplasmático, lo cual, aunque no es específico, lo orienta hacia una estirpe neuroectodérmica. Por otro lado, otra línea de investigación iniciada por Scout en 1918, describía una serie de tumores, denominados entonces neuroepiteliomas periféricos de los nervios, los cuales, Bolande en 1969, comparó con el sarcoma de Ewing y los neuroblastomas periféricos del adulto, encontrando semejanzas estructurales ópticas y electrónicas.
Actualmente, con base en los avances en los estudios anatomopatológicos, imaginológicos, inmunohistoquímicos y citogenéticos, se pone en evidencia que no nos encontramos ante un solo tumor, sino ante una familia de tumores, constituidos por células redondas, pequeñas, indiferenciadas que presentan una expresión fenotípica neuroectodérmica y una alteración genotípica común, pero con histología compleja y variable, que pueden presentarse en el tejido óseo, extra óseo u otros sistemas(3,4). Los tumores de la familia de sarcomas de Ewing (ESFT, Ewing´s Sarcoma Family of Tumors), comprende el sarcoma de Ewing clásico, sarcoma de Ewing extraóseo, tumor de Askin, sarcoma de Ewing atípico y tumor neuroectodérmico primitivo periférico (pPNET), representando un espectro que va desde las formas más indiferenciadas a las más diferenciadas, respectivamente.
Esta diferenciación se pone en evidencia mediante tinciones neurales, (como la enolasa neuroespecífica y S-4-100), o la presencia de rosetas o elementos neurales en la microscopia electrónica(2-7).
El tratamiento de estas lesiones amerita la intervención de un equipo multidisciplinario, tanto para el diagnóstico, como la implementación de las conductas terapéuticas, las cuales se basan en la cirugía, radioterapia y quimioterapia.
Debido a lo poco frecuente de este tipo de neoplasia, se cuentan con pocos reportes individualizados en la literatura; de allí, la importancia de la documentación del presente caso, en el cual, además de los hallazgos pertinentes a su origen, se suman los inherentes a su localización, crecimiento y dimensión.
CASO CLÍNICO
Se trata de adolescente masculino, de 15 años de edad, quien consultó a nuestro centro en julio de 2004, iniciando su enfermedad actual diez meses antes, en septiembre de 2003, cuando presenta nódulo redondeado en paladar duro, recubierto de mucosa, a nivel de segundo molar superior izquierdo, no doloroso; por lo cual, acude a odontólogo quien indicó antibióticoterapia por un mes sin mejoría, realizó radiografía panorámica donde se evidencia opacidad redondeada del antro maxilar izquierdo y lesión osteolítica del piso del antro, la cual es atribuida a proceso infeccioso, continuando el tratamiento (Figura 1). En vista de la persistencia de los síntomas, es llevado a centro médico privado donde realizaron en noviembre de 2003, la primera biopsia de la lesión, la cual, consistió en un fragmento de tejido en forma de disco irregular, color pardo gris, de 2,5 cm x 2 cm x 1,2 cm, la cual reportó: Fibroma con inflamación crónica y acentuada vascularización. Dos meses después aparece un nódulo redondeado con características similares al anterior, en paladar duro, a nivel del primer molar, siendo reintervenido en abril de 2004, realizando una segunda biopsia. El estudio anatomopatológico de esta muestra reportó: una pieza tisular gris blanquecina, de 2 cm x 2 cm x 1,2 cm. Los cortes histológicos muestran, a parte de proceso inflamatorio crónico inespecífico, activo, microabscedado y tejido de granulación, la presencia de células de estirpe epitelial, francamente displásicas, dispuestas en banda por debajo del epitelio de revestimiento, considerando necesario estudio inmunohistoquímico.

El estudio inmunohistoquímico (abril de 2004) reportó que el material resultó negativo a queratina y a CD45 y positivas a Kappa y Lambda, concluyendo que los hallazgos histológicos e inmunohistoquímicos son compatibles con granuloma de células plasmáticas de origen reactivo reparativo inflamatorio (lesión benigna).
Una semana después de la resección anterior aparece un nódulo no doloroso, redondeado, de crecimiento rápido, por lo cual familiares deciden consultar a la Facultad de Odontología de la Universidad Central de Venezuela en junio de 2004, momento para el cual, la tumoración tiene 4 cm de diámetro. Realizan TAC de maxilar donde se apreció lesión inflamatoria de senos paranasales y lesión redondeada, bien delimitada en antro maxilar izquierdo, que destruye el piso del mismo y se proyecta a la cavidad bucal con desplazamiento de las estructuras dentales.
Aunado al patrón de crecimiento de la lesión se suma la aparición de otra lesión posterior sésil, rojiza, aspecto irregular, por lo cual se realizó una tercera biopsia (junio de 2004) que concluye: tumor maligno de células redondas poco diferenciado.
Es entonces referido a nuestro centro, siendo ingresado al Servicio de Oncología Pediátrica del Instituto Oncológico Dr. Luis Razetti, en julio de 2004, bajo la impresión diagnóstica de tumor de células redondas poco diferenciado de maxilar izquierdo en estudio, sobre infectado.
Clínicamente el paciente se encontró febril, con asimetría facial a expensas de hemicara inferior izquierda. En cavidad bucal se apreció tumoración grande, procedente del paladar duro izquierdo, que abarca más del 50 % de la misma, sésil, granulomatosa, color rojo parduzco con extensas áreas necróticas y ulceradas, muy vascularizada, con destrucción de la arcada dentaria y desplazamiento del resto de las estructuras al lado contrario que limita la apertura bucal. Marcada pérdida del panículo adiposo. No se aprecian adenopatías. Resto del examen sin alteraciones.
Se completan estudios de extensión (TAC tórax, RMN cerebral y cara, gammagrama óseo), que demuestran lesión neoproliferativa localizada en antro maxilar y cavidad oral, sin presencia tumoral en otras áreas.
RMN cerebral y cara: (julio 2004): lesión de aspecto neoproliferativo a nivel del antro maxilar izquierdo hiper intenso en T2 heterogéneo y con iso intensidad de señal en las secuencias T1, que condiciona marcado grado de destrucción del límite óseo de la cara interna e inferior de dicho antro, con extensión del proceso hacia la región antero medial y paramedial izquierda del paladar duro, irregularmente ovoidea, poli lobulada con importante protrusión hacia la cavidad oral y destrucción del reborde alveolar del maxilar superior de ese lado. Engrosamiento mucoso inflamatorio de las celdillas etmoidales izquierdas así como del antro maxilar derecho. El resto del estudio sin imágenes patológicas ni de aspecto neoproliferativo (Figura 2).

Gammagrama óseo (julio 2004) aumento de la actividad osteoblástica en huesos de la cara maxilar superior izquierdo y derecho, sugestivo de infiltración de piso de la órbita y base del cráneo.
Se envían bloques de parafina a estudio inmunohistoquímico que reporta: CD99, ENE, cromogranina, positivo en células tumorales. Desmina, sinaptofisina, Myo-D1, Citoqueratina AE1/AE3, EMA, 34BE12, negativo en células tumorales. Hallazgos histológicos e inmunohistoquímicos compatible con pPNET.
En el curso de realización de estudios de extensión y al mismo tiempo de estabilización del paciente desde el punto de vista infeccioso, hemodinámico, respiratorio (traqueotomía) y nutricional (gastrostomía), presenta aumento persistentemente rápido de la masa, sangrado y compresión a estructuras vecinas, ocupando el 100 % de la cavidad oral e impidiendo el cierre de la misma, con un diámetro de 13 cm x 7 cm, friable y necrótico. En ese momento se indicó primer ciclo de cito reducción con VAC (Vincristina / actinomicina D / ciclofosfamida) frenando temporalmente el crecimiento. Se programa tratamiento en base a resultados inmunohistoquímicos según Protocolo SEOP-95, para sarcoma de Ewing/PNET de alto riesgo: EVAIA (Etopósido, vincristina, ifosfamida, actinomicina D, doxorrubicina), con progresión de la enfermedad, en vista de lo cual se inició radioterapia a la lesión, dosis 6 000 cGy, conjuntamente con cisplatino como radiosensibilizante. El paciente durante este período presenta pérdida de peso del 27 %, emaciación, hipotrofia muscular, hiporreflexia e hipercatabolismo que requiere intervención del equipo nutricional. Después de la radioterapia cambian las características macroscópicas de la tumoración, adquiriendo consistencia sólida, de 10 cm x 8 cm, color rosado, homogénea, que protruye por cavidad oral, con visualización de piezas dentarias inmersas en la lesión (Figura 3). El octubre de 2004 se realizó resección quirúrgica: maxilectomía bilateral con preservación del piso de la órbita (Operación de Barbosa modificada y ampliada) dejando márgenes macroscópicos libres. La evolución posoperatoria inmediata fue satisfactoria, con aparición de celulitis facial incipiente en la segunda semana, controlada con antibióticoterapia. El estudio de anatomía patológica reportó: lesión mesenquimal fibromixoide, vascularizada con áreas de mayor densidad celular perivascular, sin necrosis, mitosis ni signos de malignidad, margen posterior y arco zigomático libres de tumor, antro maxilar sin evidencia de neoplasia.
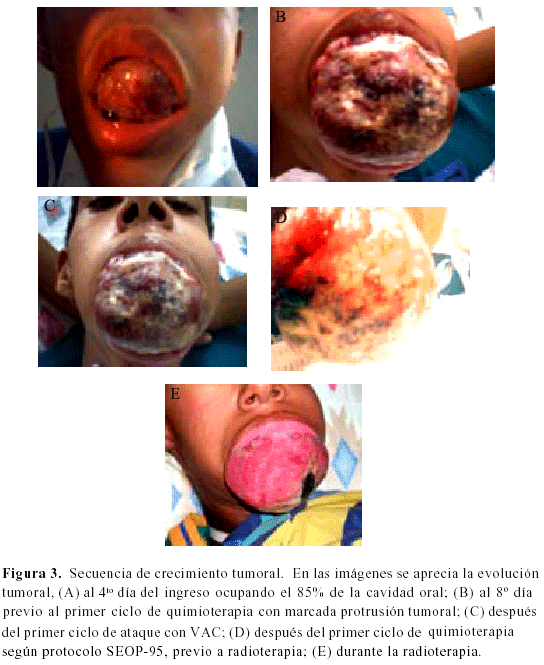
Se indicaron dos ciclos de quimioterapia con cisplatino y vincristina (dosis acumulada 520 mg/m2 sc) Continuó realizándose limpieza de detritus y secreciones por los Servicios de Cirugía de Cabeza y Cuello y Salud Bucal por dos meses con epitelización de un 70 %. Deglución disfuncional. Asimetría facial. Por atrofia muscular en miembros inferiores secundaria a desnutrición y sedentarismo, ameritó rehabilitación. Se reevaluó tres meses posterior a la cirugía con estudio de RMN: cambios posquirúrgicos regionales en la hemicara izquierda, mencionándose realce en partes blandas adyacentes al piso de la órbita izquierda (Figura 4). Gammagrama óseo: aumento de la actividad a nivel del maxilar inferior derecho y región infraorbitaria izquierda secuela del tratamiento quirúrgico. Resto del esqueleto no muestra alteraciones.

Posteriormente se tomó biopsia de tejido bucal que no mostró presencia de células tumorales. Evaluación mensual a partir de entonces sin cambios. Actualmente el adolescente se encuentra en respuesta completa, con recuperación ponderal, reintegrado parcialmente a actividades sociales y recreativas, en espera de prótesis maxilar que permita recobrar el funcionalismo de la deglución y retiro de gastrostomo (Figura 4).
DISCUSIÓN
La familia de tumores sarcomas de Ewing (ESFT), tienen una incidencia del 1 % de todos los tumores malignos y el 6 % a 11 % de los tumores óseos de la edad pediátrica. De los tumores de esta familia, el sarcoma de Ewing óseo representa el 87 %, el extraóseo el 8 % y el pPNET el 5 % de los casos. Este último, se localiza en cabeza y cuello sólo en el 6 % de los pacientes. La presentación en maxilar es sumamente infrecuente(2-5).
Los ESFT ocurren más comúnmente en la segunda década de la vida (64 %), seguido de los menores de 10 años (27 %), y la tercera década (9 %)(2-5).
La incidencia anual en EE.UU es de 2,1 casos por millón de niños. En relación al sexo, es más frecuente en el masculino en una relación de 3:2(2-5).
Clínicamente su presentación es variable, según el sitio de origen primario, extensión y la variante histológica. El 48 % de los pacientes muestran síntomas inespecíficos de aproximadamente 3 meses de evolución antes del diagnóstico, dados por pérdida de peso, dolor localizado de tipo intermitente (68 %), o continuo (96 %) y fiebre en el 21 % de los casos. Una masa palpable puede encontrarse en el 61 % de los pacientes(2-5). La hemorragia y necrosis que comúnmente acompañan al ESFT provocan aumento local de la temperatura, mimetizando procesos infecciosos, lo cual puede retrazar el diagnóstico(2). Otros hallazgos incluyen pérdida del apetito, aumento de la velocidad de sedimentación globular (VSG) y LDH, anemia y leucocitosis. La radiografía suele mostrar lesión destructiva ósea, pero es frecuente que una tumoración de tejido blando se pueda extender a las estructuras óseas adyacentes y viceversa, siendo en estos casos difícil discernir el origen de la lesión primaria(5).
En el caso presentado, una lesión en cavidad oral recurrente, con el antecedente de una radiografía panorámica que muestra radio opacidad del antro maxilar izquierdo y lesión lítica del piso del antro, orientó al cirujano a la toma de biopsia, sin embargo, ambas muestras son tomadas del tejido blando a través de la cavidad oral, sin incluir el hueso lesionado ni el tejido del antro, siendo éste el primer hallazgo radiológico observado, explicando la razón de que la muestra no fuese representativa para el diagnóstico.
Radiológicamente se muestra con lesiones líticas, escleróticas o mixtas, clásicamente denominadas en "piel de cebolla". Este efecto se debe a la formación laminada paralela de periostio en la que el tumor continuó creciendo mediante intentos de reparación del hueso. La biopsia debe ser bien planificada y, si existe sospecha de neoplasia, debe ser referido a un centro con mayor experiencia. En nuestro caso, observamos que posterior a la tercera biopsia se incrementa el tamaño tumoral, sobreinfectándose, lo que agrava el estado general del paciente. La tomografía computarizada es útil para definir la extensión hueso afectado, pero la resonancia magnética nuclear es superior para definir la extensión de la enfermedad tanto ósea como de las partes blandas, su relación con vasos sanguíneos y con órganos vecinos, así como, para el monitoreo de la respuesta a la quimioterapia.
Al presentarse una tumoración de cabeza y cuello, localizada en antro maxilar, se plantean varias posibilidades diagnósticas, orientadas por el grupo etario y epidemiología, mientras se espera el resultado de histología e inmunohistoquímica, entre las que se destacan los linfomas, rabdomiosarcomas y carcinoma nasofaríngeo. La descripción de tumor de células redondas indiferenciado conlleva a incluir los ESFT.
En la actualidad, los estudios que utilizan marcadores inmunohistoquímicos, citogenéticas, genética molecular y cultivo de tejido indican que los ESFT derivan todos de la misma célula madre primordial, es decir, células pluripotenciales que se pueden diferenciar a células con características mesenquimales, epiteliales, e incluso neurales. La hipótesis más aceptada en la actualidad es que se originan de las células colinérgicas parasimpáticas posganglionares(6,7).
La diferenciación de los ESFT de otros tumores de células redondas pequeñas de la infancia es difícil en base solo a la histología. Deben considerarse la aplicación de varios marcadores y comparar la inmunorreactividad. En el pasado, el sarcoma de Ewing típico indiferenciado era definido sólo por la inmunorreactividad para la vimentina y enolasa neuroespecífica (NSE)(7); en contraste los pPNET reaccionaban a la vimentina, así como, a los marcadores neurales (NSE, cromogranina) o acompañadas de rosetas o pseudo rosetas en el estudio morfológico.
Ambros y col.(7), en 1991, demostraron la especificidad de la expresión del gen MIC2, un gen pseudoautosómico localizado en el brazo corto de los cromosomas X y Y en los ESFT. Se han desarrollado dos anticuerpos monoclonales (12E7 y HBA-71), hoy en día designados CD99 y en ocasiones conocidas como "tinción de Ewing". Sin embargo, también puede expresarse en el tumor de Wilms no blastomatoso, sarcoma de células claras del riñón, tumores pancreáticos neuroendocrinos, teratomas inmaduros, ependimomas y tumores del plexo coroideo(6).
En el paciente encontramos positividad ante el CD99, NSE y cromogranina, lo que muestra importante diferenciación neural y negatividad a otros marcadores que descarta rabdomiosarcoma, linfoma o tumores epiteliales.
Se han promulgado varios factores predictivos, dentro de los cuales, encontramos como de mal pronóstico: metástasis al momento del diagnóstico, más de 8 cm de diámetro mayor, tumor voluminoso, localización pélvica, valores altos de LDH, y edad mayor de 17 años. Dentro de los factores de mejor pronóstico se encuentra buena reacción radiográfica a la quimioterapia de inducción o buena reacción anato-mopatológica a la quimioterapia de inducción(8,9).
Los estudios complementarios no evidenciaron enfermedad metastásica. El porcentaje de pacientes que presenta metástasis varía de una serie a otra, pero en promedio es de 25 %. Los sitios más frecuentes son: pulmón en un 38 %, hueso 31 % y médula ósea en un 11 %. Las metástasis en otros sitios, como sistema nervioso central, son raras, excepto después de la recurrencia(10).
Para la instauración de tratamiento es indispensable el diagnóstico lo más preciso posible. La meta del tratamiento de los ESFT es la curación del paciente, conservando la función y minimizando las secuelas a largo plazo. En los casos en que la curación no es realizable, el tratamiento resulta usualmente en la regresión del tumor, lo que conlleva a prolongar la sobrevida y mejorar la calidad de la misma.
En el caso que presentamos, durante la fase de estabilización del paciente se apreció una progresión acelerada de la enfermedad tumoral, por lo cual, se inició quimioterapia cito reductora, con drogas de comprobada actividad sobre los tumores probables de esa localización (Vincristina, actinomicina D y ciclofosfamida), en espera de la inmunohistoquímica(11-13). Esta combinación es activa tanto en los rabdomiosarcomas como en los ESFT. El desarrollo de protocolos de poliquimioterapia sistémica ha incrementado la supervivencia libre de enfermedad desde un 5 %-10 % en la década de los 70 a más de 70 % en la actualidad, logrando además eliminar la enfermedad diseminada microscópica y reducir el tamaño del tumor primario, facilitando su tratamiento quirúrgico. Los estudios norteamericanos IESS 1 y 2, y los europeos CESS 81 y 86, han aportado información decisiva. Los citostáticos con mayor eficacia demostrada son: vincristina, ciclofosfamida, actinomicina D, doxorrubicina, ifosfamida y etopósido. Los diferentes protocolos coinciden en dos aspectos, la estratificación de los pacientes previos al control local según el riesgo y después de la cirugía según la respuesta a la quimioterapia neoadyuvante(2,11-14).
Se decide entonces, seguir Protocolo de la Sociedad Española de Oncología Pediátrica (SEOP-95) Alto Riesgo (EVAIA) sin respuesta después de la misma(2). En ese momento debido al estado clínico del paciente, características friables de la lesión, y localización que incrementaban la morbilidad quirúrgica, se indica radioterapia. La cirugía requiere siempre de la individualización del paciente. La resección quirúrgica completa debe ser llevada a cabo en todos los casos en que se considere por el cirujano técnicamente posible y siempre tras el tratamiento quimioterápico inicial(2,15).
El tratamiento con radioterapia puede conseguir un control satisfactorio en pacientes con ESFT aunque con resultados inferiores a la cirugía. Las dosis utilizadas oscilan entre 50 y 60 Gy. Numerosos estudios demuestran una mayor supervivencia en los pacientes tratados con cirugía con o sin radioterapia, frente a los tratados con radioterapia exclusivamente. Durante el curso de radioterapia se asoció cisplatino como radiosensibilizante. El cisplatino es activo en osteosarcoma, rabdomiosarcoma y neuroblastoma, y ha mostrado actividad frente a ESFT resistente a la quimioterapia convencional.
En el caso expuesto anteriormente, la cirugía se llevó a cabo en el momento más adecuado, con cambios en las características tumorales que permitieron la resección, con márgenes microscópicos y macroscópicos libres. El estudio histológico de la masa resecada muestra un tejido fibromixoide muy vascularizado sin evidencia de tumor viable, lo que evidencia la respuesta al tratamiento.
La calidad de vida del paciente mejoró, al igual que el estado nutricional. Esto plantea una nueva perspectiva en cuanto a las secuelas de los pacientes a largo plazo, desde el punto de vista estético, factor de suma importancia en el desarrollo de los adolescentes, y el cual incluye el apoyo psicológico. El resultado hasta el momento es la integración del paciente a su entorno cotidiano, libre de enfermedad, en entrenamiento con obturador palatino y en espera de prótesis maxilar que permita deglución funcional y retiro de la gastrostomía.
En conclusión, los ESFT de localización maxilar, son pocos frecuentes. Tomando en cuenta sus factores pronósticos (tamaño, localización, metástasis al diagnóstico, niveles de LDH y respuesta a la quimioterapia inicial) debe individualizarse el tratamiento con el fin de lograr control local y erradicación de la enfermedad residual. La importancia del reporte de estos casos es el aporte de experiencias terapéuticas que contribuyan a estrategias aplicables a localizaciones complejas.
REFERENCIAS
1. Ewing J. Diffuse endothelioma of bone. Proc N Pathol Soc. 1921;21:17. [ Links ]
2. Horowitz ME, Malawer MM. Ewing´s sarcoma family of tumours. En: Pizzo P, Poplack D, editor. Principles and practice of paediatric oncology. Filadelfia: Lippincott; 1997.p.831-863. [ Links ]
3. Llombart Bosch A. Sarcoma de Ewing y tumor neuroectodérmico primitivo periférico de huesos y partes blandas. IV Congreso Virtual Hispanoamericano de Anatomía Patológica. Universidad de Valencia. España. 2004. Disponible en: URL: http://conganat.uninet.edu/IVCVHAP/CONFERENCIAS/Llombart/index.html [ Links ]
4. Mahiques A. Tumores de la familia del Ewing (EFT). 2004. Disponible en URL: http://www.arturomahiques.com/sarcoma_de_ewing.htm [ Links ]
5. Grier HE. The Ewing family of tumors. Ewings sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am. 1997;44(4):991-1004. [ Links ]
6. Parham DM, Hijazi Y, Steinberg SM, Meyer WH, Horowitz M, Tzen CY, et al. Neuroectodermal differentiation in Ewings sarcoma family of tumors does not predict tumor behavior. Hum Pathol. 1999;30(8):911-918. [ Links ]
7. Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewings sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewings sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67(7):1886-1893. [ Links ]
8. Bacci G, Picci P, Mercuri M, Ferrari S, Longhi A, Cesari M, et al. Predictive factors of histological response to primary chemotherapy in Ewings sarcoma. Acta Oncol. 1998;37(7-8):671-676. [ Links ]
9. Bacci G, Ferrari S, Longhi A, Rimondini S, Versari M, Zanone A, et al. Prognostic significance of serum LDH in Ewings sarcoma of bone. Oncol Rep. 1999;6(4):807-811. [ Links ]
10. Shankar AG, Ashley S, Craft AW, Pinkerton CR. Outcome after relapse in an unselected cohort of children and adolescents with Ewing sarcoma. Med Pediatr Oncol. 2003;40(3):141-147. [ Links ]
11. Sirvent N, Kanold J, Levy C, Dubousset J, Zucker JM, Philip T, et al. Non-metastatic Ewings sarcoma of the ribs: The French Society of Pediatric Oncology Experience. Eur J Cancer. 2002;38(4):561-567. [ Links ]
12. Evans RG, Nesbit ME, Gehan EA, Garnsey LA, Burgert O Jr, Vietti TJ, et al. Multimodal therapy for the management of localized Ewings sarcoma of pelvic and sacral bones: A report from the second intergroup study. J Clin Oncol. 1991;9(7):1173-1180. [ Links ]
13. Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewings sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694-701. [ Links ]
14. Bacci G, Mercuri M, Longhi A, Bertoni F, Barbieri E, Donati D, et al. Neoadjuvant chemotherapy for Ewings tumour of bone: Recent experience at the Rizzoli Orthopaedic Institute. Eur J Cancer. 2002;38(17):2243-2251. [ Links ]
15. Bacci G, Forni C, Longhi A, Ferrari S, Donati D, De Paolis M, et al. Long-term outcome for patients with non-metastatic Ewings sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer. 2004;40(1):73-83. [ Links ]
