










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Facultad de Ingeniería Universidad Central de Venezuela
versión impresa ISSN 0798-4065
Rev. Fac. Ing. UCV v.22 n.4 Caracas 2007
Estudio de mezclas ternarias de Poliamida-6 con Polietilenos
CARMEN ROSALES, ROSESTELA PERERA, MIREYA MATOS, YELITZA BRITO, BEATRIZ CARDOZO
Grupo de Polímeros II. Departamento de Mecánica, Universidad Simón Bolívar, Valle de Sartenejas. Apdo. Postal 89000. Baruta. Caracas. Venezuela. rperera@usb.ve
RESUMEN
En este trabajo se estudió la compatibilidad en mezclas de Poliamida-6 (PA) y diferentes polietilenos: polietileno de alta densidad (PEAD), polietileno de baja densidad (PEBD) y polietileno lineal de baja densidad (PELBD). Se utilizó un copolímero estireno-etileno/butileno-estireno funcionalizado con dietil maleato como agente compatibilizante (SEBS-f-DEM). Lacaracterización de las mezclas se llevó a cabo utilizando las técnicas de Calorimetría Diferencial de Barrido (DSC),Espectroscopía Infrarroja de Transformada de Fourier (FTIR), Microscopía Electrónica de Barrido (SEM) y la determinación de las propiedades de impacto Izod con entalla. Los ensayos de extracción selectiva realizados permitieron inferir la probable existencia de interacciones entre los grupos de la PA y del SEBS-f-DEM. Por medio de DSC se evidenció el fenómeno de cristalización fraccionada de la PA sólo en las mezclas compatibilizadas cuya fase dispersa es la PA. En general, se obtuvo un mejoramiento de las propiedades de impacto de la PA en las mezclas reactivas en las que se utilizó PELBD como matriz. Mediante el análisis por SEM de las superficies de fractura de las mezclas, se pudo evidenciar que la incorporación del SEBS-f-DEM como agente compatibilizante produce una disminución significativa en el tamaño de partículas de la fase dispersa.
Palabras clave: Poliamida, Polietileno, Mezclas reactivas, Extrusión reactiva, Funcionalización.
Study of ternary blends of Polyamide-6 with Polyethylenes
ABSTRACT
In this work, the compatibility in blends of Polyamide 6 (PA) and three different Polyethylenes, High-Density (HDPE), Low- Density (LDPE) and Linear Low-Density (LLDPE) polyethylenes, was studied. A copolymer of styrene/ethylene-butylene/ styrene functionalized with diethyl maleate (SEBS-g-DEM) was used as compatibilizing agent. The characterization of the blends was carried out using Differential Scanning Calorimetry (DSC), Fourier Transform Infrared Spectroscopy (FTIR), Scanning Electron Microscopy (SEM) and through notched-Izod impact tests. Results from selective extraction tests displayed that interactions between PA and SEBS-g-DEM groups are likely present. Through DSC results, the fractionated crystallization phenomenon of the PA was unfolded, only in those blends where the PA was the dispersed phase. In general, an enhancement in the impact resistance of those reactive blends where LLDPE was used as the matrix was obtained. SEM analyses of the fractured surfaces of the blends showed that the incorporation of SEBS-g-DEM as compatibilizing agent produced a significant decrease in the particle size of the dispersed phase.
Keywords: Polyamide, Polyethylene, Reactive blends, Reactive extrusion, Functionalization.
Recibido: diciembre de 2006 Revisado: octubre de 2007
INTRODUCCIÓN
El mezclado de polímeros permite obtener materiales con buenas propiedades físicas y procesabilidad con un amplio rango de aplicaciones en el mercado. Sin embargo, una de las principales desventajas de las mezclas de polímeros es la inmiscibilidad de la mayoría de los sistemas, debido a la naturaleza macromolecular de los componentes y a la baja interacción existente entre las fases.
Se ha encontrado que la adición de ciertas especiespoliméricas, generalmente copolímeros en bloque o eninjerto, resuelven en gran parte las desventajasanteriormente citadas que aparecen como resultado de laactividad interfacial. A estos compuestos se les denomina«agentes compatibilizantes». Tales compuestos poseensegmentos capaces de promover interacciones moleculares,como por ejemplo, puentes de hidrógeno, enlaces covalentes,interacciones dipolo-dipolo, interacciones iónicas y/oreacciones químicas entre los componentes de la mezcla a lo largo de la interfase (Akkapeddi, 2001). Mezclas de polietilenos con poliamida 6 como matriz y copolímeros funcionalizados con anhídrido maleico han sido ampliamenteestudiadas con la finalidad de aumentar la resistencia al impacto Izod con entalla de la poliamida (Thomas y Groeninckx, 1999; YU et al. 1998). Sin embargo, el uso dedietil maleato (DEM) como agente compatibilizante en estetipo de mezclas ha sido menos reportado (Sánchez et al. 2001).
En el presente trabajo se estudiaron diferentes mezclas de poliamida 6 y tres tipos de polietileno como fase dispersa(PEAD, PEBD y PELBD) en diferentes proporciones y seempleó como agente compatibilizante un estireno-etilenobutileno-estireno funcionalizado con DEM. Estas mezclas fueron caracterizadas utilizando técnicas como DSC, FTIR, SEM y mediante la determinación de las propiedades de impacto Izod con entalla.
PARTE EXPERIMENTAL
Materiales
Los polímeros estudiados en el presente trabajo fueron una Poliamida 6 (PA) Sniamid® ADS 50, grado extrusión; unpolietileno de baja densidad (PEBD) Lagotene® FB-3003 dePolímeros del Lago C.A., grado extrusión; un polietileno lineal de baja densidad (PELBD) de comonómero 1-butenoSCLAIR® 11D1 de Dupont, grado extrusión de películas; unpolietileno de alta densidad (PEAD) Altaven® 6100M de Plásticos del Lago C.A., grado extrusión de tuberías y uncopolímero tribloque (estireno-etileno-butileno-estireno) (SEBS) Kraton® G1652 lineal con una relación estireno/caucho de 29/71 de Shell Chemical Company. Se utilizótambién una mezcla de Irganox B1171 e Irganox 1098suministrados por CIBA; como estabilizantes térmicos, dietilmaleato (DEM), suministrado por Sigma Chemical Co.; comoagente funcionalizante y peróxido de dicumilo (PDC) deAldrich Chemical como iniciador de las reacciones defuncionalización. En la tabla 1 se presentan las especificaciones de los materiales utilizados.
Tabla 1. Especificaciones técnicas de los polímeros utilizados.

Funcionalización del SEBS
El SEBS fue funcionalizado con Dietil Maleato (DEM) mediante extrusión reactiva, empleando como iniciador elperóxido de dicumilo. La funcionalización se llevó a cabo enuna extrusora doble tornillo corrotante marca Berstorff,modelo ECS-2E25, bajo las condiciones de procesamientomostradas en la tabla 2. Seguidamente, el material funcionalizado fue lavado con acetona técnica.
Tabla 2. Condiciones utilizadas en la funcionalización del SEBS y en la preparación de las mezclas.

Preparación de las mezclas
Las mezclas ternarias de PA/PE/SEBS-f-DEM y las mezclasbinarias PA/PE con cada uno de los polietilenos (PEs) se prepararon en una extrusora doble tornillo corrotante marcaWerner & Pfleiderer modelo ZSK-30, según las condicionesde mezclado mostradas en la tabla 2. La cantidad de agentecompatibilizante (SEBS-f-DEM) empleado permanecióconstante para todas las mezclas (5 % del peso total de lamezcla). Se utilizaron dos estabilizantes para evitar la posibledegradación de la Poliamida (PA) durante el proceso demezclado. Estos estabilizantes fueron el Irganox B-1171 y elIrganox 1098, agregados en proporciones de 0,5 % y 0,25 %en peso, respectivamente, en base a la cantidad total de laPA presente en cada mezcla. Es importante mencionar quese empleó tanto atmósfera de nitrógeno comodesgasificación para prevenir la acumulación de humedaden la tolva y la extrusora. El extrudado fue enfriado en aguaa temperatura ambiente y luego fue granulado. Además, laPA fue secada en un horno de vacío a 80ºC por 16 horas,para evitar la hidrólisis por efecto de la humedad durante la extrusión. Las composiciones de las mezclas realizadas se presentan en la tabla 3.
Tabla 3. Composición de las mezclas PA/PE/SEBS-f-DEM.

Caracterización
Cada mezcla en estudio fue sometida a un ensayo de caracterización por Espectroscopía Infrarroja. Se obtuvieron los espectros infrarrojos de cada una de las mezclas empleando un espectrómetro infrarrojo de Transformadasde Fourier (FTIR) marca Nicolet modelo Magna 750 resolución de 4 cm-1 y señal promedio de 32 barridos. Lasmuestras se prepararon en forma de pastillas con las mezclaspulverizadas y KBr en polvo. Se prepararon dos muestraspor cada mezcla, que fueron expuestas a un intervalo desde4000 cm-1 hasta 400 cm-1 de número de onda. Además, serealizaron espectros infrarrojos del SEBS-f-DEM para determinar su grado de funcionalización. Para eliminar las diferencias por efecto del espesor de la película, se dividió la absorbancia de la banda característica del grupo carbonilodel copolímero funcionalizado a 1740 cm-1 entre la de la banda a 1601 cm-1, correspondiente a los dobles enlacesentre carbonos de los grupos aromáticos característica delSEBS, la cual, al permanecer invariable, permite determinar la cantidad de grupos carbonilos injertados en la estructura del SEBS. Para determinar la relación entre la absorbanciade los grupos carbonilos y el número de unidades del DEM injertadas, se utilizó una curva de calibración obtenida por Rosales et al. 1998.
Para verificar la posible formación de un copolímero en la interfase de las mezclas en estudio, se realizó un experimento de extracción selectiva (Anttila et al., 1999), el cual consistióen disolver muestras de 200 mg cada una de las mezclas y de los homopolímeros estudiados, en 10 ml de ácido fórmico.En aquellas mezclas en las que se observó una zonablanquecina entre el ácido y la zona insoluble se les realizó posteriormente una extracción selectiva para evaluar las posibles interacciones presentes entre los homopolímeros.
Para ello, se realizó la extracción de la poliamida soluble en ácido fórmico. Una vez separada la poliamida, se procedió a la separación del polietileno remanente colocando la muestrabajo agitación y temperatura en xileno por aproximadamente5 horas. Finalmente, para asegurar una completa separación,se realizó una extracción soxhlet de la muestra en xileno por5 horas. Para verificar la obtención de algún copolímero, seprocedió a realizar un análisis por Espectroscopía Infrarrojade Transformadas de Fourier (FTIR) al material insoluble en ambos solventes.
Se realizó la reometría dinámica a cada uno de los polímeros en estudio, en un equipo marca Rheometrics a 230ºC para la PA, el SEBS y el SEBS-f-DEM y 200ºC para los polietilenos,y a diferentes frecuencias de oscilación. También se realizó un estudio morfológico a las mezclas mediante el empleo de un Microscopio Electrónico de Barrido marca Philips, modelo505. Las muestras fueron obtenidas a partir de pequeños trozos de láminas moldeadas por compresión. Esas muestrasfueron sumergidas en nitrógeno líquido durante aproximadamente 10 minutos e inmediatamente fracturadasperpendicularmente al área transversal, con el fin de obtener una fractura frágil.
Empleando una prensa hidráulica y placas de acero recubiertas de teflón, se procedió a moldear láminas de espesor aproximado a 6 milímetros. Los ensayos deresistencia al impacto Izod con entalla de las mezclas y loshomopolímeros fueron realizados siguiendo el procedimientorecomendado en la norma ASTM D-256. Las probetas utilizadas tanto de PA como de las mezclas fueron secadaspreviamente en un horno de vacío a una temperatura de80ºC por 16 horas. Se determinaron las energías de impacto de las muestras mediante el uso de un impactómetro marca Zwick utilizando un péndulo de 1J para la PA y las mezclas80/20/5, otro de 2J para las mezclas 20/80/5 y 20/80/0, y otro de 4J para los polietilenos puros.
RESULTADOS Y DISCUSIÓN
Caracterización del agente compatibilizante
Con el fin de verificar la presencia del grupo éster proveniente del DEM injertado en el SEBS, se realizó unacaracterización del material funcionalizado mediante laTécnica de Espectroscopía Infrarroja (FTIR). En la figura 1 se presentan los espectros infrarrojos del SEBS y del SEBSf- DEM. Se observa una banda a 1740 cm-1en el espectro delSEBS funcionalizado asociada principalmente a estiramientos del grupo carbonilo (C=O) proveniente del grupo éster del Dietil Maleato (DEM), que está injertado enla estructura química del SEBS, mientras que la débil señalobservada para el SEBS sin funcionalizar se atribuye a losgrupos carbonilos generados por la degradación del materialdurante el proceso de extrusión y a los sobretonosaromáticos propios del estireno (Nakanishi, 1970). A partirde los espectros infrarrojos se obtuvieron relaciones deabsorbancia. Estas relaciones fueron realizadas entre labanda ubicada a 1740 cm-1 correspondiente a los carbonilos y la banda a 1601 cm-1 correspondiente a los dobles enlaces entre carbonos de los grupos aromáticos característica del SEBS. El grado de funcionalización del SEBS funcionalizadofue obtenido a partir de una curva de calibración reportada por Rosales et al. 1998, encontrada para SEBS funcionalizados con diferentes proporciones de DEM. En la tabla 4 se presentan las relaciones de absorbancia (A1740/ A1601) para el SEBS puro y funcionalizado y el grado defuncionalización del SEBS-f-DEM. El bajo valor obtenidoestá asociado a la alta viscosidad del SEBS y a la migración del DEM en la extrusión reactiva (Rosales et al. 1998).

Figura 1. Espectros infrarrojos del SEBS y del SEBS-f-DEM.
Tabla 4. Relaciones de absorbancia (RA), grados de funcionalización (GF), viscosidad dinámica (η*) y módulo de almacenamiento (G′) a 1 rad/s y 230°C, del SEBS, SEBS-f-DEM y de la PA.

Propiedades reológicas de los componentes de las mezclas
En las figuras 2 y 3 se presentan las curvas de viscosidad de corte dinámica en función de la frecuencia de oscilación ymódulo de almacenamiento (G´) en función del módulo de pérdidas (G´´) para los componentes de las mezclas. Lascurvas de viscosidad en función de la velocidad de cortereflejan el carácter pseudoplástico de estos materiales. Porotra parte, las curvas del módulo de almacenamiento enfunción del módulo de pérdidas son independientes del pesomolecular promedio en peso y de la temperatura parahomopolímeros y mezclas miscibles de polímeros. Lascaracterísticas reológicas obtenidas de los polietilenosestán asociadas a su estructura molecular (peso molecularpromedio en peso, distribución de pesos moleculares y lapresencia de ramificaciones largas en el caso del PEBD). Enla figura 2 se observa una menor variación de la viscosidadcon la frecuencia de oscilación para el PELBD, debido a suestrecha distribución de pesos moleculares. Además, estematerial presenta un menor módulo elástico (G´) por lacarencia de ramificaciones largas y por su estrechadistribución de pesos moleculares en comparación a la del PEBD (Fetters et al. 1994).

Figura 2. Viscosidad dinámica (η*) en función de la frecuencia de oscilación (ω) de la PA, el SEBS y el SEBS-f-DEM a 230°C, y de los polietilenos a 200°C.

Figura 3. Módulo de almacenamiento (G′) en función del módulo de pérdidas (G″) de la PA, del SEBS y del SEBS-f-DEM a 230°C, y de los polietilenos a 200C.
Por otra parte, en el SEBS funcionalizado (SEBS-f-DEM) se obtiene una menor viscosidad dinámica y un menor módulode almacenamiento (figuras 2 y 3). Las diferencias observadasentre el SEBS y el material funcionalizado son un indicativo de reacciones secundarias de escisiones de cadena en elproceso de extrusión reactiva. Estas reacciones secundariasproducen una reducción en la viscosidad y le confieren almaterial menor rigidez en el fundido, es decir, menor módulode almacenamiento a las temperaturas de procesamiento(Rosales et al. 1998). De todos los componentes de lasmezclas, la PA es la que posee la menor viscosidad de corte y módulo de almacenamiento a altas frecuencias de oscilación.
Morfología de las mezclas
En el mezclado en extrusoras, el tamaño de las partículas dela fase dispersa está influenciado por los flujos de corte quetienden a romper las partículas, los flujos elongacionalesque las deforman antes de romperlas y el efecto deaglomeración de las mismas (coalescencia) que se favorecepor la inmiscibilidad de las mezclas y por la cantidad de partículas dispersas (Wu, 1987 y Akkapeddi, 2001). En lapreparación de mezclas reactivas, además del grado defuncionalización hay que tomar en cuenta las viscosidadesy la elasticidad en fundido de los componentes a la velocidadde deformación del proceso de mezclado y la migración del agente compatibilizante a la superficie de la fase dispersa (efecto emulsificante) que se favorece en matrices de baja viscosidad (Thomas y Groeninckx, 1999). Todos estos factores afectan la morfología de las mezclas.
En las figuras 4 a 7 se muestran las micrografías de las mezclas. La diferencia esencial entre las mezclas binarias y ternarias estudiadas es el menor número de huecos que se También es apreciable la reducción en el tamaño de las partículas en estas mezclas compatibilizadas. En las mezclas no reactivas de composición 20/80 (sin agentecompatibilizante, figuras 4a y 4b), el menor tamaño departículas se obtiene para la mezcla con PEAD como matriz. Se hubiese esperado el menor tamaño de partículas en lasmezclas no reactivas realizadas con el PELBD como matriz,debido a su mayor viscosidad a la velocidad de deformacióndel proceso de mezclado (alrededor de 100 s-1). Sin embargo,hay que tomar además en cuenta el menor módulo elásticodel fundido del PELBD, que reduce la probabilidad dedeformación de las partículas de PA dispersadas en él. Poresta razón, se obtienen mayores tamaños de partículas en las mezclas con PELBD. En cuanto al PEBD, aunque éste posee menor viscosidad de corte, su mayor módulo dealmacenamiento (mayor rigidez en el fundido, es decir mayorelasticidad) comparado con el del PEAD y el del PELBD secontrapone al efecto viscoso y por ende permite la obtenciónde tamaños de partículas similares a los de las mezclasrealizadas con PEBD y PELBD como matrices (Vanoene, 1972 y Meijer y Janssen, 1994).

Figura 4. Micrografías de las mezclas no reactivas de composición 20/80; (a): PA/PEAD y (b): PA/PEBD.

Figura 5. Micrografías de las mezclas reactivas de composición 20/80/5; (a): PA/PEAD/SEBS-f-DEM y (b): PA/PEBD/SEBS-f-DEM.

Figura 6. Micrografías de las mezclas reactivas; (a): PA/PELBD/SEBS-f-DEM, 20/80/5 y (b): PA/PELBD/SEBS-f-DEM, 80/20/5.

Figura 7. Micrografías de las mezclas reactivas de composición 80/20/5; (a): PA/PEAD/SEBS-f-DEM y (b): PA/PEBD/SEBS-f-DEM.
En cuanto a las mezclas reactivas de composición 20/80/5 cuya fase dispersa es la PA (figuras 5a y 5b), además de losparámetros reológicos hay que tomar en cuenta la reducciónde la tensión interfacial que se produce por el efecto del agente compatibilizante y la migración de éste hacia lasuperficie de la fase dispersa (efecto emulsificante). En el proceso de mezclado hay dos efectos reológicoscontrapuestos, por lo que no se observan diferencias apreciables en el tamaño de partículas de las tres mezclas(figuras 5a, 5b y 6a). Es de hacer notar que en la mezclareactiva cuya fase continua es el PELBD y la fase dispersa PA6/SEBS-f-DEM (figura 6a) se observa adhesión entre las fases (puntos de conexión) y además, hay cierta aglomeración de las partículas más pequeñas y una separación muy clara en la interfase de los componentes.
Las micrografías de las mezclas reactivas de composición 80/20/5 cuya matriz es la PA se muestran en las figuras 6b, 7a y 7b. Para estas mezclas, el menor tamaño de partículas seobserva en la que tiene PELBD como fase dispersa, debido a su menor módulo de almacenamiento en comparación alos otros polietilenos (figura 3). En el proceso de mezcladola probabilidad de rompimiento de las partículas es mayorcuando su elasticidad en el fundido es menor (Vanoene, 1972). Por otra parte, el tamaño de las partículas de las fases dispersas es mayor en las mezclas reactivas cuya matriz esla poliamida (figuras 6b, 7a y 7b) debido a la menor viscosidady elasticidad en el fundido de la PA y por ende, a los menoresesfuerzos elongacionales y de corte a que se someten las partículas de cada polietileno en el proceso de mezclado.
Caracterización de las mezclas mediante FTIR y extracción selectiva
En el estudio de las mezclas mediante la técnica de FTIR seobservaron diferencias entre los espectros de las mezclas yel espectro de la poliamida 6. Entre estos cambios seencuentra el desplazamiento de la banda asociada a los carbonilos libres hacia menores frecuencias, así como ladisminución de la intensidad de la misma banda y un aumento de la banda asociada a los carbonilos enlazados. Sin embargo, estas variaciones espectrales no son indicativasde una posible interacción de la PA6 y el agente compatibilizante, ya que las diferencias antes mencionadas también se observan en las mezclas no reactivas. Paraverificar la naturaleza de las interacciones entre la PA y elDEM injertado en el SEBS, se realizó un ensayo desolubilidad de las mezclas en ácido fórmico. Esta prueba serealizó a todas las mezclas, a los homopolímeros y al agente compatibilizante. La PA se disolvió completamente en ácido fórmico, ya que este polímero es soluble en el solvente. Lospolietilenos y el SEBS-f-DEM permanecieron en estado sólido, comprobando así su insolubilidad en el ácido.
Entre las observaciones realizadas se tiene que las mezclas ternarias PA6/PE/SEBS-f-DEM de composición 20/80, tanto reactivas como no reactivas, presentan partículas insolubles blancas flotando en la superficie de una solución incolora,mientras que las mezclas PA/PE/SEBS-f-DEM de composición 80/20/5 mostraron una turbidez ubicada en la interfase entre la solución incolora y las partículas blancasinsolubles, luego de una inmersión por aproximadamente 24horas. La aparición, incremento y persistencia de turbidez,que representa una suspensión coloidal de partículas, seproduce debido a la posible existencia de copolímeros deinjerto que actúan como agentes interfaciales (Anttila et al. 1999; Yu et al. 1998 y Scaffaro et al. 2003).
Debido a la dificultad para caracterizar la fase turbia intermedia encontrada en la disolución de las mezclasreactivas con ácido fórmico, se procedió a realizar unaextracción selectiva de la fracción de poliamida 6 que noreaccionó con el grupo éster injertado en el SEBS y de lafracción olefínica. Para ello, las mezclas PA/PE/SEBS-f-DEMde composición 80/20/5 fueron colocadas en ácido fórmicopor 48 horas con el fin de eliminar la fase de PA, yposteriormente colocadas en xileno a 230ºC por 10 horas,para eliminar la fracción de PE y los restos de SEBS-f-DEMpresentes en la muestra. Por medio de esta extracciónselectiva se pretendió verificar la presencia de copolímerode injerto, formado durante el mezclado, producto de las probables interacciones entre los grupos N-H de la PA y los grupos éster del SEBS-f-DEM. El material obtenido luego de la extracción selectiva fue caracterizado por Espectroscopía Infrarroja (FTIR). Debido a que no seapreciaron cambios significativos en las bandas ubicadasen la región entre 3000-3500 cm-1, el estudio espectroscópico se realizó en la región comprendida entre 1500-1800 cm-1.
En la figura 8 se muestra el espectro infrarrojo de la mezclaPA/PEAD/SEBS-f-DEM antes y después de la extracción,así como el espectro correspondiente a la PA en la regióncomprendida entre 1500-1800 cm-1. En esta figura se observa la presencia de un hombro o lomo ubicado entre 1710 cm-1 y 1750 cm-1 en el espectro, luego de la extracción, a diferencia de los espectros de la PA y de la mezcla, antes de la extracción. Esta diferencia podría ser indicio de la existencia deinteracciones entre los grupos N-H de la PA y el carbonilodel DEM presente en el SEBS funcionalizado, lo cual traeríacomo consecuencia la formación de un copolímero de injerto. En los espectros de las mezclas, antes de la extracción, no aparece el hombro ubicado entre 1710 cm-1 y 1750 cm-1,debido probablemente a un efecto de solapamiento por partede la banda ubicada a 1645 cm-1. Es de hacer notar que se encontraron resultados similares en las mezclas con PEBD y PELBD después de la extracción (no se muestran las figuras).

Figura 8. Espectros infrarrojos en la región entre 1500-1800 cm-1 de la PA, del material resultante de la extracción con ácido fórmico y xileno y de la mezcla sin extracción PA/PEAD/SEBS-f-DEM de composición 80/20/5.
En resumen, mediante el ensayo de extracción selectiva se infiere la formación de un copolímero de injerto PA-SEBS-f-DEM, el cual actúa como agente interfacial en las mezclasPA/PE/SEBS-f-DEM de composición 80/20/5 para todos los polietilenos utilizados. Realizando una extracción selectivaen estas mezclas para clarificar la naturaleza de lasinteracciones se puede inferir la formación de copolímeros de injerto, debido a la presencia de una banda que podríaestar asociada al grupo succinimida. Sin embargo, se creeque este copolímero se forme en bajas proporciones, debidoa que la banda asociada a este grupo sólo se observó en ladeconvolución de los espectros de las mezclas luego de la extracción. Además, no se pueden descartar interaccionesde amidación que puedan ocurrir entre el grupo éster queforma parte del agente funcionalizante y el grupo terminal amino. Estas pruebas sólo se realizaron a las mezclas mayoritarias en PA debido a la mayor proporción de grupos N-H2.
Propiedades térmicas de las mezclas
En las tablas 5 y 6 se presentan las propiedades térmicas de los materiales puros y de sus mezclas. Tanto los termogramas de fusión como los de cristalización de las mezclaspresentaron los picos correspondientes a las fases presentesde PA y al tipo de polietileno utilizado (PEAD, PELBD y PEBD). Estos resultados parecen indicar que, independientemente de la composición de la mezcla, lospolímeros son inmiscibles en estado fundido, ya que lospicos de fusión de cada componente aparecen de maneradefinida e independiente, a temperaturas muy cercanas alas de los polímeros puros, dentro del rango del error experimental de los ensayos.
Tabla 5. Temperaturas de fusión (Tf pico)y de cristalización (Tc pico) de los componentesde las mezclas PA/PE/SEBS-f-DEM de diferentes composiciones, medidas en el segundo calentamiento.

Tabla 6. Temperaturas de cristalización pico (Tc pico) y entalpías de cristalización (ΔHc) de la fase PA en las mezclas PA/PE/SEBS-f-DEM de composiciones diferentes.

Por otra parte, en los termogramas de calentamiento de las mezclas se presentaron variaciones en la endoterma de fusiónasociada a la PA, como consecuencia de una posible reducción de las estructuras cristalinas inestables «γ» aladicionar el SEBS-f-DEM. Estas diferencias podrían seratribuidas a interacciones entre el agente compatibilizante ylos homopolímeros (Anttila et al. 1999). Sin embargo, no seobservaron cambios apreciables en las entalpias de fusióny cristalización de las mezclas con PA como fase mayoritaria.Resultados similares han sido obtenidos en otros estudioscon diferentes tipos de polietilenos como fase dispersa (Yaoet al. 2000; Halldén et al. 2001; Minkova et al. 2002; Jiang et al. 2003; Scaffaro et al. 2003).
En la figura 9 se presentan los termogramas de enfriamiento de las mezclas reactivas PA/PEBD/SEBS-f-DEM cuya fase dispersa es la PA (composiciones 20/80/5, 35/65/5 y 50/50/5,respectivamente). En estas figuras pareciera notarse la presencia de un tercer pico de cristalización alrededor de133°C (400 K). Este comportamiento también se observó en las mezclas con PELBD de composición 20/80/5 y 35/65/5,es decir, las mezclas donde la PA es la fase minoritaria, y esreportado por Frensch et al., 1989, como cristalizaciónfraccionada en la poliamida. Este fenómeno no se observóen las mezclas no reactivas ni en las mezclas reactivas con PEAD como fase mayoritaria.
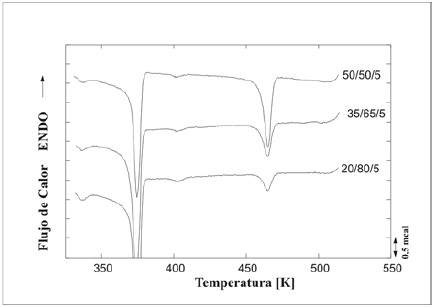
Figura 9. Termogramas de enfriamiento de las mezclas reactivas PA/PEBD/SEBS-f-DEM cuya fase dispersa es la PA.
La cristalización fraccionada está relacionada con la capacidad de un material de cristalizar a diferentes temperaturas, especialmente a menores temperaturas conrelación a su temperatura de cristalización estándar. Esta capacidad está ligada a las características del proceso denucleación, es decir, dependiendo de la activación de losnúcleos presentes en la muestra. Cuando el material seencuentra disperso en muchas partículas pequeñas delorden de las micras, el número de heterogeneidades puedeser menor que el número de partículas de material, por loque la nucleación en estos microdominios no ocurrirá de la manera heterogénea dominante en el polímero en masa,requiriéndose de mayores sobreenfriamientos para que otrasheterogeneidades se activen o que el propio polímero formenúcleos (nucleación homogénea). En las mezclas PA6/PEBD/SEBS-f-DEM y PA6/PELBD/SEBS-f-DEM que presentan cristalización fraccionada, la PA está dispersa en un gran número de gotas muy pequeñas, por lo que no en todas existirán heterogeneidades que permitan que la nucleaciónocurra heterogéneamente. Por lo tanto, se requerirá desobre enfriamientos mayores y la nucleación tenderá a ocurrir preferencialmente de forma homogénea. Según este razonamiento se esperaría que aquellas gotas que sí presentan heterogeneidades (que son por lo general restos de catalizador remanente del proceso de polimerización), actúen como núcleos y que parte del material cristalice aaltas temperaturas, mientras que la otra parte del material,donde las gotas no presentan heterogeneidades, cristalicea temperaturas menores, lo cual parece estar ocurriendo en las mezclas en las cuales la PA es la fase dispersa.
La cristalización no se ve afectada por el mezclado cuando la PA es el componente mayoritario en la mezcla, pero cuando es el componente minoritario cambia su conducta cristalina, apareciendo en el enfriamiento dos exotermas claramente identificables. La exoterma a mayor temperatura (192°Caproximadamente) corresponde a la temperatura usual decristalización de la poliamida 6, mientras que la exoterma a menor temperatura (alrededor de 117°C K y 142°C) corresponde a la formación de cristales por efecto de la activación de algún segundo tipo de heterogeneidad quedefine la cristalización fraccionada. Este planteamiento secorrobora con los resultados obtenidos en el estudio demicroscopia electrónica de barrido (SEM), donde el tamañode partículas de la PA6 presente en las mezclas PA6/PE/ SEBS-f-DEM de composición 20/80/5 es mucho menor ypresenta una mejor dispersión en comparación con lasmezclas 80/20/5. Moon et al. 1991, al estudiar mezclas de PPcon PA6 compatibilizadas con PP-f-MAH, encontraron que la adición del agente compatibilizante reduce la temperaturay reduce también, de manera significativa, la exoterma de cristalización de la fase minoritaria de PA6. Estas observaciones sugieren que el compatibilizante, bajointeracciones específicas con la PA6, provoca cambios enla cinética de cristalización de la fase dispersa, es decir, el compatibilizante interfiere en la estructura cristalina de laPA6 y por tanto decrece la velocidad de cristalización. En el presente estudio se evidencia la existencia de cambios en la cinética de cristalización de las mezclas donde la PA6 es la fase dispersa y se emplea SEBS-f-DEM como agente compatibilizante, cambios que pueden ser atribuidos al efecto que el compatibilizante ejerce sobre la PA6.
En los termogramas de enfriamiento de las mezclas PA6/ PEAD de composición 20/80 con y sin agente compatibilizante se observó una diferencia significativa enel pico de cristalización correspondiente al PEAD, ya que la mezcla sin compatibilizante presentó dos picos en laexoterma similares a los que se observaron en el PEAD puro, mientras que la mezcla con compatibilizante presenta unsolo pico de cristalización bien definido (tabla 5). Al revisarlas temperaturas correspondientes a cada uno de los picos, se tiene que el primer pico se encuentra a 117°C y el segundoa 121°C en las mezclas 20/80/0; mientras que el pico de lamezcla con compatibilizante se ubica a 119°C. Esto sugiere que ambas fracciones cristalinas sufren un mezclado oreordenamiento que podría atribuirse a la presencia delSEBS-f-DEM. Sin embargo, no se puede asegurar que este cambio en la cristalización del PEAD sea debido al compatibilizante, pues no se ha descartado que este cambiopodría también ocurrir en las mezclas PEAD/SEBS y/o en las mezclas PA6/PEAD/SEBS.
Estudio de las propiedades de impacto Izod con entalla
La poliamida 6 es un polímero que exhibe un comportamiento frágil y el polietileno, por su parte, es un polímero dúctil. En la figura 10 se presentan los valores de resistencia al impacto Izod con entalla obtenidos para los homopolímeros y lasmezclas PA/PE/SEBS-f-DEM. En esta figura se observa que el PEAD posee menor resistencia al impacto que el PELBD yéste a su vez menor al PEBD. Este hecho es consecuenciade la cristalinidad de estos materiales. Los cristales poliméricos actúan como enlaces o entrecruzamientos y producen una reducción en la resistencia al impacto Izodcon entalla. En general, las mezclas cuya matriz es polietileno poseen mayor resistencia al impacto, que aquellas en dondeconstituye la fase dispersa, debido al comportamiento dúctil que caracteriza a estas poliolefinas.

Figura 10. Resistencia al Impacto Izod con entalla (RI) en función de la composición de las diferentes mezclas.
Los valores de la resistencia al impacto Izod con entalla de las mezclas, cuya fase mayoritaria es la PA, están por debajo de una ley aditiva de mezclas. La disminución en la resistenciaal impacto observada en estas mezclas podría ser causadapor la poca adhesión entre la matriz y la fase dispersa de las mezclas, por lo que las partículas que forman la fase dispersa actuarían como concentradores de esfuerzos que facilitaríanla formación y propagación de la grieta en la matriz frágil de la PA. En el mezclado reactivo, aunque se obtuvo evidencia de interacciones entre los componentes de las mezclas por la extracción selectiva y FTIR, no se obtuvo el tamaño promedio óptimo de partículas para alcanzar altos valores en esta propiedad en aquellas mezclas con PA como fasemayoritaria. Este tamaño óptimo de partículas es de 0,3 μmpara mezclas de poliamida 6 con cauchos y menor paramezclas con polietilenos (Akkapeddi, 2001; Aróstegui et al. 2001).
Por otra parte, el valor de resistencia al impacto para la mezcla PA/PELBD/SEBS-f-DEM de composición 20/80/5 está porencima de la ley aditiva de mezclas. Esta mayor resistenciase debe a su morfología y ésta, a su vez, es consecuencia delas diferencias en contracciones térmicas de los diferentesmateriales empleados. En las otras mezclas con PA comofase dispersa, tanto reactivas como no reactivas, los valoresse encuentran por debajo. La diferencia esencial entre las mezclas con los diferentes tipos de polietilenos es lamorfología de las mismas, aunque el tamaño de las partículasde PA es similar en las tres mezclas, como se dijo conanterioridad. En las mezclas con el PELBD como fase mayoritaria (figura 6a) se observa una gran separación entre las partículas de PA y la matriz (PELBD), lo que podríadisminuir la propagación de la grieta en los ensayos deimpacto. Kim et al. 1998 desarrollaron un modelo del proceso de la deformación micromecánica, para explicar la altaductilidad encontrada en mezclas de PP/PA/SEBS-f-AM con PA como fase dispersa. En este modelo se presenta, en primer lugar, una dilatación de las partículas debido a laconcentración de esfuerzos. En el caso de partículasdilatadas de PA y/o aglomeradas (figura 6a), los esfuerzos triaxiales en un ensayo de impacto producen huecos en lainterfase entre fase dispersa y matriz que se propagan y producen bandas de cizallamiento con el consiguiente aumento de la resistencia al impacto Izod con entalla.
CONCLUSIONES
Mediante el análisis por SEM de las superficies de fractura de las mezclas, se pudo evidenciar que la incorporación delSEBS-f-DEM como agente compatibilizante produce unadisminución significativa en el tamaño de partículas de la fase dispersa. Los ensayos de extracción selectiva realizadospermitieron inferir la probable existencia de interaccionesentre los grupos de la PA y del SEBS-f-DEM y que el probable copolímero de injerto PA-SEBS-f-DEM actúa como agente interfacial en las mezclas PA/PE/SEBS-f-DEM decomposición 80/20/5 para todos los polietilenos utilizados.
Mediante la técnica de DSC se verificó la incompatibilidadde los homopolímeros empleados, puesto que todas lasmezclas presentan las endotermas de fusión asociadas a lapoliamida 6 y a los polietilenos. Además, se evidenció elfenómeno de cristalización fraccionada de la PA sólo en las mezclas compatibilizadas cuya fase dispersa es la PA. Lasmezclas PA6/PEAD presentan dos picos en la exoterma decristalización asociadas al PEAD al igual que el PEAD puro, mientras que las mezclas PA6/PEAD/SEBS-f-DEM presentan un solo pico.
En general, se obtuvo un mejoramiento de las propiedades de impacto de la PA en las mezclas reactivas en las que se utilizó PELBD como matriz. Sin embargo, los valores de laresistencia al impacto Izod con entalla de las mezclas cuyafase mayoritaria es la PA están por debajo de una ley aditivade mezclas. Aunque se obtuvo evidencia de interaccionesentre los componentes de las mezclas por la extracción selectiva y FTIR, no se obtuvo el tamaño promedio óptimo de partículas para alcanzar altos valores en esta propiedad.
REFERENCIAS
1. AKKAPEDDI, K. (2001). Rubber toughening of polyamides by reactive blending. Reactive Polymer Blending. Eds. Baker W., Scott C., Hu G.-H., Hanser. Munich. p. 208. [ Links ]
2. ANTTILA, U., HAKALA, K., HELAJA, T., LÖFGREN, B. AND SÉPALA, J. (1999). Compatibilization of polyethylene/polyamide 6 blends with functionalized polyethylenes preparedwith metallocene catalyst. J. Poly. Sci., Part A: Polym.Chem. 37; pp. 3099-3107. [ Links ]
3. ARÓSTEGUI, A., GAZTELUMENDI, M. AND NAZÁBAL, J. (2001). Toughened poly(butylenes terephtalate) by blending with a mettallocenic poly(ethylene-octene) copolymer.Polymer 42; pp. 9565-9574. [ Links ]
4. FETTERS, L. J., LOHSE, D. J., RICHTER, D., WITTEN, T. A. AND ZIRKEL, A. (1994). Connection between polymermolecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 27 (17); pp. 4639-4647. [ Links ]
5. FRENSCH, H., HARNISCHFEGER, P. AND JUNGNIICKEL, B. (1989). Fractionated crystallization in incompatible polymer blends en Multiphase polymers: blends and ionomers. Eds. L. A. Utraki y R. A. Weiss, American Chemical Society. pp. 101. [ Links ]
6. HALLDÉN, A., DERISS, M. J. AND WESSLÉN, B. (2001). Morphology of LDPE/PA-6 blends compatibilized withpoly(ethylene-graft-ethylene oxide). Polymer 42; pp. 8743-8751. [ Links ]
7. JIANG, C., FILIPPI, S. AND MAGAGNINI, P. (2003). Reactive compatibilizer precursors for LDPE/PA6 blends. II:maleic anhydride grafted polyethylenes. Polymer 44; pp. 2411-2422. [ Links ]
8. KIM, G. M., MICHLER, G. H., RÖSCH, J. AND MÚLHAUPT, R. (1998). Micromecanical deformation processes in toughened PP/PA/SEBS-g-MA blends prepared by reactiveprocessing. Acta Polymer. pp. 49; 88-95. [ Links ]
9. MAGAGNINI, P. (2003). Reactive compatibilization of PA6/ LDPE blends with an ethylene-acrylic acid copolymerand a low mass bis-oxazoline. Polymer. 44; pp. 6951- 6957. [ Links ]
10. MEIJER H. & J. JANSSEN, (1994). Mixing of immiscible liquids en Mixing and compounding of polymers. Eds. Manas I. y Tadmor Z. Hanser Publications. Munich. p. 85. [ Links ]
11. MINKOVA, L., YORDANO, H. R. AND FILIPPI, S. (2002). Characterization of blends of LDPE and PA6 withfunctionalized polyethylenes. Polymer 43; pp. 6195- 6204. [ Links ]
12. MOON, H., RYOO, B. AND PARK, J. (1991). Concurrent crystallization in polypropylene/nylon-6 blends using maleic anhydride grafted polypropylene as acompatibilizing agent. J. Poly. Sci., Part A: Polym. Phys. 32; pp. 1427-1434. [ Links ]
13. NAKANISHI, K. (1970). Infrared absorption spectroscopypractical. Holden-Day eds. p. 14. [ Links ]
14. ROSALES, C., PERERA, R., ROJAS, H., MEJÍA, K., SÁNCHEZ, A. (1998). Viscoelastic and morphological study of polyamide-6/polyethylene/SEBS-g-DEM ternary blends. J. Macrom. Sci., Pure Appl. Chem. A35 (7 & 8); pp. 1187-1205. [ Links ]
15. SÁNCHEZ, A., ROSALES, C., LAREDO, E., MÜLLER, A. J. AND PRACELLA, M. (2001). Compatibility studies in binary blends of PA6 and ULDPE-graft-DEM. Macromol. Chem.Phys. 202; pp. 461-2478. [ Links ]
16. SCAFFARO, R., LA MANTIA, F., CANFORA, L., POLACCO, G. ANDFILIPPI, S. (2003). Reactive compatibilization of PA6/LDPE blends with an ethylene-acrylic acid copolymerand a low molar mass bis-oxazoline. Polymer 44; pp.6951-6957. [ Links ]
17. THOMAS, S., GROENINCKX, G. (1999). Reactive compatibilization of heterogeneous ethylene propylene rubber (EPM)/ nylon 6 blends by the addition of compatibiliser precursor EPM-g-MA. Polymer 40; pp. 5799-5819. [ Links ]
18. VANOENE, H. (1972). Modes of dispersion of viscoelastic fluids in flow. J. Colloid and Interface Sci. 40 (30); pp. 448-467. [ Links ]
19. WU, S. (1987). Formation of dispersed phase in incompatible polymer blends: interfacial and rheological effects. Polym. Eng. Sci. 27; pp. 335-341. [ Links ]
20. YAO Z., Z. YIN, G. SUN, C. LIU, J. TONG AND L. REN. (2000). Morphology, thermal behavior, and mechanical properties of PA6/UHMWPE blends with HDPE-g- MAH as a compatibilizing agent. J. Appl. Polym. Sci. 75; pp. 232-238. [ Links ]
21. YU Z. Z., Y. C. OU. AND G. H. HU. (1998). Influence of interfacial adhesion on toughening of polyethylene-octene elastomer/nylon 6 blends. J. Appl. Polym. Sci. 69; pp. 1711-1718. [ Links ]
