










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. v.6 n.3 Mérida oct. 2008
Disfunción beta pancreática
Miguel Angel Contreras Zambrano*
Centro Médico El Valle, Nueva Esparta, Venezuela.
* Dirigir correspondencia a: Dr. Contreras M. miguelac@c-com.net.ve
Resumen
La disfunción beta pancreática es una realidad fisiopatológica que conduce a una pérdida gradual en la eficacia de los opciones terapéuticas utilizadas en la DM. La evidencia actual establece que tanto en los diabéticos como en la condición de prediabetes, la disfunción de la célula beta es el primer defecto demostrable con limitación de la capacidad de compensación en presencia de resistencia a la insulina. Cada vez toma más importancia la necesidad de mantener el control glucémico a largo plazo utilizando opciones terapéuticas que aseguren la preservación del funcionamiento de la masa beta pancreática.
Palabras clave: Disfunción célula beta, masa pancreática.
Abstract
The dysfunction pancreatic beta is a physiopatological reality that conducts to a gradual loss in the efficacy of the therapeutic options utilized in the DM. The current evidence establishes that so much in the diabetic as in the condition of prediabetes, the dysfunction of the cell beta is the first demonstrable defect with limitation of the capacity of clearing in the presence of resistance to the insulin. Each time takes more importance the need to maintain the control glucémico long-term utilizing therapeutic options that assure the preservation of the operation of the pancreatic mass.
Key words: Beta cell dysfunction, pancreatic mass
Artículo recibido en: Marzo 2008. Aceptado para publicación en: Junio 2008.
El deterioro progresivo de la función beta pancreática en la Diabetes Mellitas tipo 2 (DM2) fue señalado por el estudio UKPDS (Estudio Prospectivo de la Diabetes en el Reino Unido), el cual determinó que independientemente de la terapéutica utilizada (dieta, insulina, sulfonilúreas o metaformina) se produce un deterioro progresivo del control glucémico. Uno de los hallazgos más relevantes de este estudio fue que después de 6 años de monoterapia con sulfonilúreas, el 62% de los pacientes con un valor de HOMA-b < 27% (como expresión de secreción de insulina) requirieron medicamentos adicionales para lograr un mejor control de la glucemia. En contraste, solo 28% de los pacientes con HOMA-b por debajo de 55% requirieron terapia adicional1.
Relación entre resistencia a la insulina y función de las células-b
La evidencia actual establece que tanto en los diabéticos como en la condición de prediabetes, la disfunción de la célula beta es el primer defecto demostrable con limitación de la capacidad de compensación en presencia de resistencia a la insulina sin embargo, la sensibilidad de la insulina tiene un efecto modulador sobre la función de la célula beta. La naturaleza de esta relación es tan evidente, que la sensibilidad a la insulina y la función de la célula-2 están inversa y proporcionalmente relacionadas, y el producto de estos dos parámetros es una constante, denominada Índice de Disposición. Este índice puede interpretarse como una medida de la habilidad de la célula-b para compensar la resistencia a la insulina1.
Masa de células beta pancreática
La composición del páncreas adulto está determinada por un 99% de tejido exocrino constituido por microacinos que drenan sus secreciones en el ducto exocrino y un 1% de tejido endocrino formado por aproximadamente 1 millón de islotes de Langerhans. Cada islote humano contiene aproximadamente 3000 células-b que tienden a agruparse en el centro del acino2.
Regulación de la masa de células-b pancreática
La masa beta pancreática es regulada por tres mecanismos independientes:
a. Replicación: división mitogénica de las células pre-existentes.
b. Neogénesis: formación de nuevas células a partir de las células ductales epiteliales preexistentes.
c. Apoptosis: muerte celular programada.
La suma de los índices de replicación y neogénesis de las células beta menos el índice de apoptosis nos da el índice total de crecimiento de la masa beta3 (Figura 1).

Evolución de la masa de células-b pancreática
El tamaño de la masa-b del adulto es determinado por su desarrollo durante la embriogénesis y la niñez. Los niños tienen tasa de replicación alta, la cual disminuye en forma importante después de la infancia temprana. No existen datos que apoyen la neogénesis como fuente de crecimiento en los humanos5. El periodo de incremento rápido de la masa beta durante la gestación y la infancia es vulnerable a factores nutricionales, ambientales y genéticos. El desarrollo insuficiente de la masa beta durante la gestación y la infancia puede predisponer al desarrollo de la Diabetes4,5. Finalmente, en los pacientes diabéticos se describe un aumento de hasta tres (3) veces en la tasa de apoptosis de las células beta pancreáticas4,6.
Relación entre la célula-b, el envejecimiento y la diabetes
Si utilizamos la primera fase de la secreción de la insulina como medida de la función pancreática, encontramos que en individuos no diabéticos se produce un deterioro de este parámetro a razón de un 0,7% anual, y que en individuos con intolerancia a la glucosa, dicho parámetro se deteriora a un ritmo de 1,25% anual. Al momento del diagnóstico de diabetes, el paciente ya tiene un deterioro del 80% en este marcador7. Con respecto a la masa pancreática, ésta disminuye hasta en un 40% en los individuos con hiperglucemia en ayunas y en un 60% en los pacientes diabéticos7,8 (Figura 2). La sensibilidad a la insulina per-se no disminuye con la edad y las alteraciones que se producen en ella son secundarias a cambios en la composición corporal y en la actividad física1,7.

Fisiopatología
Existen múltiples mecanismos implicados en la destrucción de la célula-b en la DM2, incluyendo la gluco-lipotoxicidad y los efectos de los productos adipocitarios (adipoquinas) inflamatorios, que originan resistencia a la insulina y aumento de la demanda secretoria de la célula-2 9. El incremento de la demanda metabólica origina múltiples señales apoptóticas que convergen en dos (2) vías ejecutorias comunes denominadas: Disfunción Mitocondrial y Estrés del Retículo Endoplásmico10.
Disfunción mitocondrial
El incremento del metabolismo de la glucosa y de los ácidos grasos libres por medio de la oxidación mitocondrial determina generación de radicales libres (ROS) y disminución de la producción de ATP. La caída en la generación de ATP compromete la secreción de insulina y a su vez potencia el estrés del retículo endoplásmico, con la apoptosis consiguiente10.
Estrés del retículo endoplásmico
El retículo endoplásmico es una organela altamente dinámica con un rol central en la biosíntesis de lípidos y proteínas. Su funcionamiento está altamente determinado por el aporte energético mitocondrial (Figura 3) y cuando es sometido a sobrecargas metabólicas con déficit de sustrato (ATP) se induce la activación de señales de apoptosis a través del mecanismo de estrés del retículo endoplásmico11.

El incremento de la demanda metabólica produce, no solo aumento en la producción de insulina; también aumenta la síntesis de proinsulina lo cual favorece el estrés del retículo endoplásmico, potenciando así la disfunción de la célula beta1.
El denominado Péptido Amiloide (IAPP) se encuentra coexpresado con la insulina en la célula beta, y se secretan simultáneamente. Este péptido inhibe directamente la secreción de insulina. El estrés del retículo endoplásmico estimula la liberación del IAPP, lo cual puede exceder su capacidad de solubilidad y con ello, facilita su transformación en oligomeros tóxicos que desecadenan respuestas apoptóticas12.
Estrategias farmacológicas: el reposo metabólico
Como se planteó anteriormente, la excesiva sobrecarga metabólica sobre la célula beta puede inducir disfunción y apoptosis a través de mecanismos donde están implicadas la mitocondria y el retículo endoplásmico13,14. Una estrategia lógica sería favorecer el descanso necesario de la célula beta (reposo metabólico) para así protegerla contra las agresiones del entorno corporal. Como ejemplo, se ha propuesto el uso de esquemas con insulina, tempranos e intensivos, que inducen remisión temporal de la disfunción15.
En este contexto, las sulfonilúreas inducen mejoría inicial en la función de la célula beta por reducir la carga de la glucotoxicidad. Posteriormente, al producir estimulación crónica de la célula, acelerarían la velocidad de pérdida de la masa beta, con la disfunción consiguiente. Es importante resaltar, que las sulfonilúreas aumentan las concentraciones de proinsulina13. La metaformina puede disminuir la demanda secretoria de insulina y, a su vez, reduce los niveles de apoptosis al disminuir el estrés oxidativo9.
Las hormonas incretinas (GLP-1 y GIP) son péptidos liberados por el tracto gastrointestinal en respuesta a la ingesta de nutrientes y tienen tres (3) tipos de efectos sobre la célula beta: agudo: potenciación de la secreción de insulina dependiente de glucosa, sub-agudo: estimulación de la trascripción del gen de la insulina con aumento de su biosíntesis y crónico: estimulación de la proliferación de las células beta con inhibición de la apoptosis e inducción en la neogénesis a partir de células ductales precursoras16.
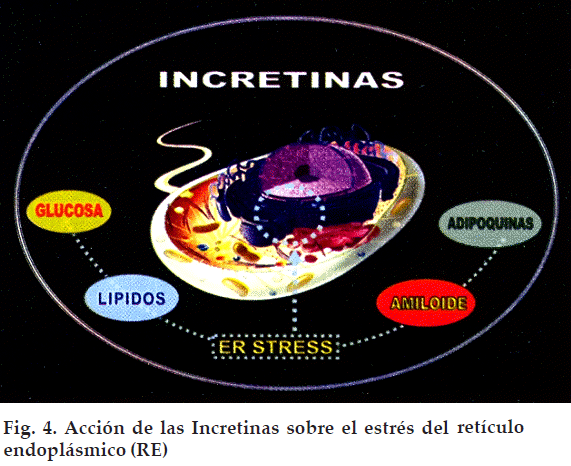
Desde el punto de vista estructural, el punto básico de acción de las incretinas es reducir el estrés del retículo endoplásmico (Figura 4). Debemos hacer notar que los efectos anteriormente señalados fueron reportados en animales de experimentación y actualmente están en desarrollo los estudios para demostrar el efecto de esta modalidad terapéutica en el control glucémico a largo plazo1.
Las Tiazolidinedionas (TZD) protegen a la célula beta de los efectos de la gluco-lipotoxicidad en forma simultánea: favorecen la disminución de los niveles de glucosa y el secuestro adipocitario de los ácidos grasos circulantes1. Además, optimizan el perfil secretor adipocitario: bloquean la producción de citoquinas inflamatorias, como el factor de necrosis tumoral alfa (TNFa) y facilitan la producción de citoquinas protectoras como la adiponectina1 (Figura 5). Desde el punto de vista estructural, las tiazolidinedionas optimizan la función mitocondrial y favorecen su biogénesis por estimular la vía reguladora del consumo energético conocida como AMPK (Proteina Cinasa Activada por AMP). En cuanto a los marcadores funcionales, las TZD reestablecen la primera fase de la secreción de la insulina en los pacientes diabéticos, disminuyen la secreción de proinsulina (aún en pacientes bajo tratamiento con sulfonilureas) y bloquean la producción del péptido amiloide1.

Todo lo anteriormente planteado se resume en dos (2) estudios clínicos: DREAM (Evaluación de la Reducción del Riesgo para Desarrollar Diabetes con Rosiglitazona) y ADOPT (Ensayo sobre la Progresión de la Diabetes) (Figura 6). El estudio DREAM seleccionó 5300 pacientes mayores de 30 años con prediabetes, para recibir tratamiento con rosiglitazona por un promedio de 3 años. La conclusión final fue una reducción de un 60% en la incidencia de DM2 con el aumento consiguiente en la probabilidad de regresión a la normoglucemia17. En el estudio ADOPT18 se seleccionaron 4300 pacientes con diagnóstico reciente de DM2 y se evaluaron 3 estrategias terapéuticas de monoterapia: sulfonilúreas (glibenclamida), metaformina y tiazolidinedionas (rosiglitazona) por un periodo de 4 años. Los pacientes que recibieron rosiglitazona mostraron una reducción del 63% en el riesgo de desarrollar falla terapéutica comparado con glibenclamida, y de un 34% comparado con metaformina. Se demostró que el efecto protector de la rosiglitazona se debió esencialmente a la acción sensibilizadora de la insulina y en menor grado, a aumento en la secreción de insulina. La metaformina mostró un efecto intermedio tanto en el aspecto sensibilizador como en el efecto secretor. La sulfonilúrea mostró el peor perfil en cuanto a preservación de su efecto terapéutico en el tiempo19.

Conclusiones
1. La disfunción beta pancreática es una realidad fisiopatológica que conduce a una pérdida gradual en la eficacia de los opciones terapéuticas utilizadas en la DM.
2. La disfunción beta pancreática es un proceso que comienza a desarrollarse muchos años antes de que hagamos el diagnóstico de DM según los criterios actuales.
3. Cada vez toma más importancia la necesidad de mantener el control glucémico a largo plazo utilizando opciones terapéuticas que aseguren la preservación del funcionamiento de la masa beta pancreática.
Referencias bibliográficas
1. Wajchenberg B. Beta- Cell failure in diabetes and preservation by clinical treatment. Endocr Rev 2007; 28:187-218. [ Links ]
2. Buttler P. Life and death of beta-cells in the islets of Langerhans in type 2 Diabetes. http://www.d4pro.com/idm/site/life and death of beta cells. [ Links ]
3. Rhodes, C. Type 2 Diabetes- a matter of b cell life and death?. Science 2005; 21;307(5708):380-384. [ Links ]
4. Buttler P. The replication of b cells in normal physiology in disease and for therapy. Nat Clin Pract Endocrinol Metab 2007;3:758-768. [ Links ]
5. Buttler P. Beta cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008;57:1584- 1594. [ Links ]
6. Buttler P. 2-cell deficit and increased 2 cell apoptosis in humans with type 2 Diabetes. Diabetes 2003;52:102-110. [ Links ]
7. Gerich J. Effect of aging on glucose homeostasis. Diabetes Care 2008;31:539-543. [ Links ]
8. Festa A. Beta-cell dysfunction in subjects with impaired glucose tolerance and early type 2 diabetes. Comparison of surrogate markers with first phase insulin secretion from an intravenous glucose tolerance test. Diabetes 2008;57:1638-1644. [ Links ]
9. Standl E. The importance of b-cell management in type 2 diabetes. Int J Clin Pract Suppl 2007;153:10- 19. [ Links ]
10. Prentki M. Islet 2 cell failure in type 2 diabetes. J Clin Invest 2006;116:1802-1812. [ Links ]
11. Eizirik D. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008;29:42-61. [ Links ]
12. Butler P. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev 2008;29:303-316. [ Links ]
13. Aston-Mourney K. Too much of a good thing: why it is bad to stimulate the beta cell to secrete insulin. Diabetologia 2008;51:540-545. [ Links ]
14. Nichols CG. 2-cell hyperexcitability: from hyperinsulinism to diabetes. Diabetes Obes Metab 2007;9 Suppl 2:81-88. [ Links ]
15. Li Y. Induction of long term glycemic control in newly diagnosed type 2 diabetic patients is associated with improvement of 2-cell function. Diabetes Care 2004; 27:2597-2602. [ Links ]
16. Perfetti R. The role of GLP1 in the regulation of islet cell mass. Horm Metab Res 2004;36:804-810. [ Links ]
17. Gerstein HC. Effects of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomized controlled trial. Lancet 2006;368:1096-1105. [ Links ]
18. Kahn S. Glycemic durability of rosiglitazone, metformin or glyburide monotherapy. N Engl J Med 2006;355:2427-2443. [ Links ]
19. Goldstein Barry. Clinical Translation of A Diabetes Outcome Progression Trial: ADOPT Appropriate Combination Oral Therapies in Type 2. J Clin Endocrinol Metab 2007;92:1226-1228. [ Links ]

