










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. v.8 n.8 Mérida feb. 2010
SISTEMA RENINA ANGIOTENSINA Y RIESGO CARDIO-METABÓLICO.
Marcos M. Lima1, José Carmelo Nuccio2, Marjorie Villalobos1, Christopher Torres3, Nathalie Balladares3
1Unidad de Endocrinología. Instituto Autónomo Hospital Universitario de los Andes.
2Centro de Investigaciones Cardiológicas. Instituto Autónomo Hospital Universitario de los Andes.
3Laboratorio de Estudios Cardiovasculares y Neurociencias. Universidad de Oriente. Núcleo Bolívar. Dirigir correspondencia a: Dr. Marcos M. Lima. marcoslimamedical@hotmail.com
RESUMEN
En años recientes el concepto clásico del sistema renina angiotensina ha experimentado cambios sustanciales. La identificación de los nuevos componentes del sistema renina angiotensina ha contribuido a modificar nuestro entendimiento acerca de su función en condiciones fisiológicas así como en diversas enfermedades. En este artículo de revisión nos enfocaremos en el papel de este sistema endocrino en el riesgo cardiometabólico.
Palabras Clave: Sistema renina angiotensina, angiotensina II, hipertensión, diabetes.
ABSTRACT
In the past few years the classical concept of the reninangiotensin system has experienced substantial changes. The identification of the new components of the reninangiotensin system have contributed to switch our understanding about its function in physiological situations and in several diseases. In this review we will focus on the role of this endocrine system in the cardiometabolic risk.
Key Words: Renin-angiotensin system, angiotensin II, hypertension, diabetes.
Artículo recibido en: Diciembre 2009. Aceptado para publicación en: Enero 2010.
El Sistema Renina Angiotensina (SRA) es considerado un sistema endocrino cuyos metabolitos activos tienen una amplia variedad de funciones en diferentes órganos y tejidos. En la circulación, una proteasa altamente específica denominada renina, es capaz de convertir el angiotensinógeno de origen hepático en un decapéptido la angiotensina I (Ang I), el cual a su vez es convertido a angio-tensina II (Ang II) por acción de la enzima convertidora de angiotensina (ECA)1 Esta enzima se encuentra altamente expresada en las membranas de las células endoteliales de la circulación pulmonar y tiene la capacidad de inactivar también al sistema de las bradicininas2.
En años recientes, el concepto del SRA ha experimentado cambios sustanciales, uno de ellos es el descubrimiento del SRA tisular o local, el cual se caracteriza por la presencia de los componentes del SRA a nivel de los tejidos3. En el año 1971 Ganten y cols.4 demostraron por primera vez que los componentes del SRA podían ser producidos localmente a nivel de varios órganos y tejidos, y este SRA local parece ser regulado independientemente del SRA circulante, pero también puede interactuar con este último. De esta manera, los efectos del SRA local pudieran ocurrir en las mismas células que producen los péptidos (efecto intracrino y autocrino), en células vecinas (efecto paracrino) o a través de la circulación a órganos o tejidos específicos3,5.
Este nuevo entendimiento del SRA ha permitido una mejor comprensión del efecto de este complejo sistema endocrino en varios órganos, así como su participación en un gran número de patologías que pueden ocurrir como consecuencia de la sobreactividad de este sistema. En este artículo trataremos la relación que existe entre el SRA y el riesgo cardiometabólico.
COMPONENTES DEL SISTEMA RENINA ANGIOTENSINA Angiotensinógeno
El angiotensinógeno es una glucoproteína de 452 aminoácidos producido en el hígado, así como en otros tejidos incluyendo el corazón, riñones y tejido adiposo, el cual circula como un péptido biológicamente inactivo. Por medio de la acción de la renina, el angiotensinógeno es convertido en Ang I, el cual constituye el péptido precursor del SRA1.
Renina
La renina es una proteasa producida por las células del aparato yuxtaglomerular del riñón y es considerada una enzima clave del SRA debido a la naturaleza limitante de su actividad hidrolítica sobre el angiotensinógeno1. En años recientes la renina ha adquirido mayor importancia debido al descubrimiento del receptor de prorenina/renina (RPR, del inglés renin prorenin receptor). El RPR es un receptor transmembrana expresado en grandes cantidades en las células mesangiales, corazón, cerebro, adipocito visceral y en las células del músculo liso vascular6,7.
La (pro)renina representa del 70 al 90% de la renina circulante en sujetos normales y más del 95% en pacientes con diabetes mellitus6,7. La (pro)renina es un zimógeno catalíticamente inactivo que se une al RPR e induce un incremento en la conversión catalítica de angioten-sinógeno a Ang I. Además, la unión de la (pro)renina a su receptor genera una cascada de señales intracelulares asociadas con la activación de la proteincinasa asociada a mitógenos (MAPK, del inglés mitogenactivated protein kinase), la cinasa reguladora de señales extracelulares tipo 1 y 2 (ERK 1/2, del inglés extracellular signal-regulated kinases) y la fosforilación de la proteína de choque térmico 27 (HSP27, del inglés heat shock protein 27), conllevando a un aumento en la síntesis de ADN, colágeno tipo 1, fibronectina y factor de crecimiento transformador β-1 (TGF-β1, del inglés transforming growth factor- β-1), los cuales son conocidos como mediadores en procesos de fibrosis y remodelado en varias enfermedades8-10 (Fig. 1). Estos descubrimientos han abierto las puertas a un nuevo grupo de medicamentos inhibidores directos de la renina.
Enzima Convertidora de Angiotensina
El papel de la ECA dentro del SRA está bien establecido desde los trabajos pioneros de Skeggs y cols. en el año 195611, los cuales demostraron que la ECA constituía la enzima clave en la generación de Ang II. Cuarenta y dos años después, Deddish y cols12 describieron la acción de la ECA en el catabolismo de Angiotensina (1-7). De esta manera, la ECA es capaz de producir un potente vasoconstrictor la Ang II e inactivar a la Ang (1-7) que tiene efectos vasodilatadores al actuar sobre el receptor Mas.

Fig. 1. Acciones Bioquímicas de la (Pro)Renina sobre su Receptor
En el año 2000, dos grupos independientes identificaron una nueva enzima homóloga de la ECA, a la cual denominaron enzima convertidora de angiotensina 2 (ECA2)13,14. Esta enzima es homóloga en un 42% con la ECA, pero con actividades bioquímicas diferentes. La ECA 2 al hidrolizar a la Ang I genera Angiotensina (1-9), la cual sirve como una vía indirecta para generar Ang II; sin embargo, la actividad catalítica de la ECA2 es 400 veces mayor sobre la Ang II que sobre la Ang I, y conlleva a la formación de Ang (1-7) con propiedades vasodilatadoras como se mencionó anteriormente13,14. De esta manera, el SRA puede ser visto como un sistema endocrino dual en el que las acciones vasoconstrictoras/proliferativas y las acciones vasodilatadoras/antiproliferativas son reguladas en parte por un balance entre la ECA y la ECA2, lo cual hace fácilmente entendible el efecto benéfico que tienen los inhibidores de la enzima convertidora de angiotensina (IECA) en el perfil de pacientes cardiometabólicos.
Angiotensina II
La Ang II fue aislada por primera vez en 1940 por Braun-Menendez y cols15 y en un principio fue caracterizada como un potente vasoconstrictor que incrementa la resistencia vascular periférica y en consecuencia eleva la presión arterial. En situaciones de depleción del volumen extracelular la Ang II reduce la excreción renal de sodio y agua alterando la hemodinámica renal y además estimula la secreción de aldosterona por la corteza suprarrenal la cual provoca mayor reabsorción hidrosalina a nivel del túbulo contorneado distal y del túbulo colector. De esta manera la Ang II regula la presión arterial de forma directa al aumentar la resistencia vascular periférica y de forma indirecta al aumentar el volumen sistólico y por ende el gasto cardíaco16.
El receptor celular de la Ang II fue identificado en 1974 como un receptor de membrana con alta afinidad por la Ang II17. Posteriormente, en el año 2000 se identificaron dos subtipos de receptores: los AT1 y los AT218. En humanos, los receptores AT1 son ampliamente expresados en los vasos sanguíneos, corazón, riñón, glándulas suprarrenales e hígado y los AT2 están presentes principalmente en tejidos fetales, disminuyendo rápidamente después del nacimiento, encontrándose en baja cantidad en los tejidos de los adultos. Los receptores AT1 median los efectos ya señalados de la Ang II; mientras que los AT2 median efectos opuestos como vasodilatación, antiproliferación celular y apoptosis19. Este conocimiento ha permitido el desarrollo de bloqueadores de los receptores AT1 de Ang II (BRA); sin embargo el bloqueo de los receptores AT1, así como la inhibición de la ECA, son capaces de estimular el asa de retroalimentación de la renina, la cual como ya se comentó tiene efectos vasoconstrictores y proliferadores intrínsecos (Fig. 2)20,21
EL SISTEMA CARDIOVASCULAR EN EL SINDROME METABÓLICO
Más de un tercio de las muertes ocurridas a nivel mundial pueden ser atribuidas a un pequeño grupo de factores de riesgo, donde los cinco principales en orden decreciente son: hipertensión arterial, tabaquismo, hiperglicemia, sedentarismo y obesidad22. Se estima que al menos 50% de los pacientes hipertensos son resistentes a la insulina, siendo ésta una anormalidad fundamental en la patogénesis del síndrome metabólico o cardiometabólico23.
El incremento de la adiposidad visceral se asocia con alteraciones del metabolismo de la glucosa y lípidos, e hipertensión arterial, no habiéndose demostrado la misma correlación con la adiposidad subcutánea24. El vínculo entre adiposidad visceral y complicaciones cardiometabólicas se enfoca en la interrelación entre la sensibilidad a la insulina, el sistema nervioso simpático, el SRA y el sistema de péptidos natriuréticos cardiacos25.
CAMBIOS EN EL SISTEMA CARDIOVAS-CULAR ASOCIADOS A LA OBESIDAD
La obesidad se caracteriza por un incremento del volumen plasmático y del gasto cardiaco con valores de resistencia vascular periférica en rango normal. Los obesos hipertensos muestran un aumento de la resistencia vascular periférica cuando se comparan con obesos normotensos26. Los obesos además, responden a la hipertensión con un patrón geométrico del ventrículo izquierdo predominantemente excéntrico, exhibiendo mayor tendencia a la insuficiencia cardiaca, con un aumento del riesgo de 5% para hombres y 7% para mujeres por cada unidad de incremento del índice de masa corporal27.
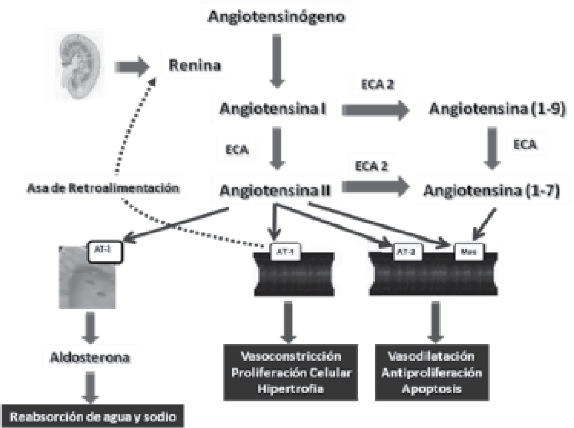
Fig. 2. Sistema Renina Angiotensina Aldosterona
SISTEMA RENINA ANGIOTENSINA, SISTEMA NERVIOSO SIMPÁTICO Y PÉPTIDOS NATRIURÉTICOS
Los pacientes con obesidad visceral tienen niveles elevados de todos los componentes del SRA a pesar de un incremento en la ingesta de sodio, retención hidrosalina e hipertensión arterial. Se ha demostrado que la dieta hipocalórica y la pérdida de peso es capaz de corregir los niveles séricos y la actividad de la renina plasmática, angiotensinógeno y aldosterona, por lo que se infiere que la dieta hipercalórica juega un papel preponderante en la activación de este sistema25.
La regulación anormal del SRA en pacientes obesos puede deberse a un incremento primario en los componentes del sistema o una falta de inhibición por parte de los sistemas antagonistas como los péptidos natriuréticos cardiacos. El adipocito es capaz de generar cada uno de los componentes del SRA, de manera tal que el incremento de la adiposidad visceral conlleva a un aumento en la producción local de angiotensinógeno y por ende a mayor actividad del SRA25. La expresión genética del angiotensinógeno en el adipocito es distinta a la hepática, y parece estar regulada por la alimentación. Una pérdida de 5 % de peso corporal se asocia con una disminución de la expresión genética y de los niveles circulantes de angiotensinógeno28.
Por otro lado, se ha documentado la hiperactividad del sistema nervioso simpático en el síndrome metabólico y por ende en el estado de resistencia a la insulina. Los efectos simpaticoexcitatorios de la resistencia a la insulina están mediados por una acción central, a través de un efecto facilitador de la insulina sobre la actividad simpática a nivel del hipotálamo ventromedial, lo cual crea un ambiente favorable para el desarrollo de hipertensión arterial29,30. Además, la leptina es capaz de mediar la activación del sistema simpático renal a través de la vía de la fosfatidil inositol - 3 cinasa (PI3K, del inglés phospha-tidylinositol 3-kinase), enzima responsable de las acciones metabólicas de la insulina31. La estimulación simpática constituye el principal determinante de la secreción de renina desde la mácula densa, y puede además aumentar la expresión de angiotensinógeno en los adipocitos, haciendo retroalimentación positiva con el SRA. De la misma manera, la angiotensina II puede incrementar el tono simpático directamente a nivel central, o a nivel periférico facilitando la transmisión post-sináptica e inhibiendo la recaptación de noradrenalina23.
Tanto el péptido natriurético auricular como el ventricular pueden inhibir directamente la secreción de renina y aldosterona, así como al sistema simpático y la secreción de vasopresina. Igualmente, inhiben la proliferación de preadipocitos a adipocitos maduros, siendo este tejido el segundo lugar después del riñón en poseer la mayor cantidad de receptores para estas moléculas32,33. Sin embargo, el sistema de los péptidos natriuréticos se encuentra disminuido en pacientes con síndrome metabólico, a pesar del incremento del tamaño ventricular. Esto es consecuencia de un aumento de su depuración en el tejido adiposo y un desacoplamiento del receptor y su segundo mensajero34,35. Actualmente el fenómeno de inhibición de los péptidos natriuréticos cardíacos es visto como el primer elemento que ocurre en la hipertensión arterial asociada al síndrome metabólico, y la hiperactividad sostenida del SRA y del sistema nervioso simpático, es secundaria a la falta de inhibición por este sistema.
ANGIOTENSINA II Y DIABETES MELLITUS TIPO 2
Las acciones de la Ang II sobre la sensibilidad insulínica han sido descritas en múltiples estudios desde que la resistencia a la insulina es considerada como un factor de riesgo independiente para el desarrollo de enfermedades cardiovasculares como hipertensión arterial y aterosclerosis36.
La unión de la insulina a las subunidades α del receptor de insulina, lleva consigo la activación de la tirosina cinasa con la consiguiente autofosforilación de los residuos de tirosina del sustrato del receptor de insulina (IRS, del inglés insulin receptor substrate), esto producirá a su vez la activación de la cascada fisiológica de la PI3K y la posterior inducción de procesos metabólicos como la glucogénesis, glucólisis, síntesis de proteínas y lipogénesis. Se ha propuesto que la fosforilación de los residuos de serina/treonina del IRS bloquea la vía de la PI3K e induce resistencia a la insulina activando la cascada de la MAPK la cual tiene importantes efectos mitogénicos y proliferadores37.
Khamzina y cols. describieron que la activación insulínica del blanco en mamíferos de la rapamicina (mTOR, del inglés mammalian target of rapamycin) y la cinasa ribosomal S6 tipo 1 (S6K-1, del inglés ribosomal S6 kinase-1 ) disminuyen la sensibilidad a la señal PI3K de la insulina en hepatocitos, posiblemente por incremento de la fosforilación de los residuos de serina del IRS, comportándose esta vía por tanto como un mecanismo de retroalimentación negativa que regula las señales insulínicas en diferentes tejidos38.
En años recientes se ha descrito que los receptores AT1 están conectados a vías de señalización usualmente asociadas con factores de crecimiento y receptores de citoquinas. Esto ocurre principalmente a través del acopla-miento del receptor AT1 a la transactivación del Factor de Crecimiento Epidérmico (EGF, del inglés epidermal growth factor) el cual media eventos celulares tales como crecimiento, proliferación y migración celular16,39. Estudios han demostrado que la Ang II al actuar sobre su receptor AT1 produce activación de metaloproteasas de matriz (MMP, del inglés matrix metalloproteases) las cuales liberan EGF que activa a su vez a su receptor (EGFR, del inglés epidermal growth factor receptor), lo cual conlleva a la activación de la ERK 1/2, que posteriormente activa la mTOR/S6K-1 que producen fosforilación de los residuos de serina del IRS con la posterior desensibilización de la señal PI3K de la insulina, induciendo así a través de este complejo mecanismo resistencia a la insulina (Fig. 3)39,40.
Por su parte, en el páncreas endocrino la actividad intrínseca del SRA regula el flujo sanguíneo dentro del islote y permite el reconocimiento de niveles elevados de glucosa y la oportuna liberación de insulina y de otras hormonas pancreáticas y péptidos mediadores. Estas observaciones surgen de estudios en roedores, en los cuales se evidenció que mejora el flujo sanguíneo a los islotes cuando reciben un IECA o un BRA. Estos cambios en la perfusión del páncreas pueden explicar el retraso en la primera fase de liberación de insulina en respuesta a la glucosa (disfunción de la célula beta); sin embargo, los mecanismos intrínsecos de este efecto aún no están completamente dilucidados41-43.
Adicionalmente, la Ang II puede afectar el número y función de las células beta ya que incrementa el estrés oxidativo, la apoptosis y fibrosis del páncreas. Cuando la Ang II actúa sobre sus receptores AT1 es un poderoso estímulo para la formación de radicales libres de oxígeno en los vasos sanguíneos por activación de las oxidasas de nicotinamida adenina dinucleótido reducido (NADH) y nicotinamida adenina dinucleótido fosfato reducido (NADPH) que se encuentran aumentadas en estados de hiperglicemia, esto favorecerá la producción de anión superóxido y de peróxido de hidrógeno, así como la formación de productos avanzados de glicosilación, lo cual conllevará a glucotoxicidad de la célula beta con la posterior apoptosis de la misma44,45 . Asimismo, la Ang II incrementa la producción de citoquinas y factores de crecimiento fibrogénicos que contribuyen a la aparición de placas de amiloide dentro del islote46.

Fig. 3. Resistencia a la Insulina inducida por Angiotensina II
Estos conocimientos fisiopatológicos se han visto reflejados en la práctica clínica dado que la inhibición de la ECA ha demostrado mejorar la sensibilidad a la insulina y el control glicémico en pacientes con diabetes así como una reducción del 14% de riesgo relativo en la incidencia de nuevos casos de diabetes según datos del estudio de prevención con Captopril47. Se ha postulado que este efecto benéfico se logra a través de dos mecanismos principales: un efecto hemodinámico, ya que favorece el flujo sanguíneo a nivel de la microcirculación de tejidos sensibles a la acción insulínica, principalmente tejido adiposo, músculo esquelético y célula beta-pancreática, lo cual facilitaría la acción de la insulina a la vez que promueve su secreción y además mejora la acción de la insulina a nivel de su receptor disminuyendo las concentraciones de Ang II y las alteraciones que ésta produce en la vía de señalización de la insulina48. En pacientes diabéticos, el bloqueo del SRA tiene efectos cardioprotectores, al disminuir la rigidez arterial (perindopril, valsartán, losartán), la disfunción diastólica ventricular (candesartán, quinapril, losartán), la progresión de la calcificación de las arterias coronarias, la disfunción endotelial inducida por hiperglicemia, así como también la respuesta inflamatoria (Valsartán)49.
Actualmente se lleva a cabo el estudio NAVI-GATOR, acrónimo del inglés Nateglinide and Valsartan in Impaired Glucose Tolerance Out-comes Research, con seguimiento a 7 años, donde se evalúa diabetes mellitus de nuevo inicio y aparición de eventos cardiovasculares mayores, a fin de demostrar si el bloqueo del SRA previene el desarrollo de diabetes mellitus50. Por otro lado, están surgiendo medicamentos novedosos que permiten
intervenir de manera positiva en el balance del SRA, tal es el caso del uso de ECA2 recombinante humana en modelos de ratones diabéticos, lo cual ha demostrado que es capaz de reducir la progresión de la nefropatía diabética mediante la disminución de la presión arterial y de la actividad de las oxidasas de NADPH, lo cual a todas luces refleja el efecto protector de la ECA2 y su posible uso a futuro para reducción de riesgo cardiometabólico global51.
REFERENCIAS BIBLIOGRÁFICAS
1. Zaman MA, Oparil S, Calhoum DA. Drugs targeting the renin-angiotensin aldosterone system. Nature 2002; 1: 621-636. [ Links ]
2. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev 2003; 24: 261-271. [ Links ]
3. Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86: 747-803. [ Links ]
4. Ganten D, Marquez-Julio A, Granger P, Hayduk K, Karsunky KP, Boucher R, Genest J. Renin in dog brain. Am J Physiol 1971; 221: 1733-1737. [ Links ]
5. Miyazaki M, Takai S. Tissue angiotensin II generating system by angiotensin-converting enzyme and chymase. J Pharmacol Sci 2006; 100: 391-397. [ Links ]
6. Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Intern Med 2008; 264: 224-236. [ Links ]
7. Nguyen G. Renin/prorenin receptors. Kidney Int 2006; 69: 1503-1506. [ Links ]
8. Jan Danser AH, Batenburg WW, Van Esch JH. Prorenin and the (pro) renin receptor –an update. Nephrol Dial Transplant 2007; 22: 1288-1292. [ Links ]
9. Nguyen G, Burckle C, Sraer JD. The renin receptor: the facts, the promise and the hope. Curr Opin Nephrol Hypertens 2003; 12: 51-55. [ Links ]
10. Batenburg WW, Jan Danser AH. The (Pro) renin receptor: a new addition to the renin-angiotensin system?. Eur J Pharmacol 2008; 585: 320-324. [ Links ]
11. Skeggs LT, Kahn JR, Shumway NP. The preparation and function of the hyperten-converting enzyme. J Exp Med 1956; 103: 295-299. [ Links ]
12. Deddish PA, Marcic B, Jackman HL, Wang HZ, Skidgel RA, Erdös EG. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin (1-7) and keto-ACE. Hypertension 1998; 31: 912-917. [ Links ]
13. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart R, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000; 87: E1- E9.
14. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner A. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000; 275: 33238-33243. [ Links ]
15. Braun-Menendez E, Fasciolo JC, Leloir LF. The substance causing renal hypertension. J Physiol (Lond) 1940; 98: 283-298. [ Links ]
16. Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 2006; 20: 953-970. [ Links ]
17. Glossmann H, Baukal A, Catt KJ. Angiotensin II receptors in bovine adrenal cortex. Modification of angiotensin II binding by guanyl nucleotides. J Biol Chem 1974; 249: 664-666.
18. De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger Th. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 2000; 52: 415-472. [ Links ]
19. Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 2000; 52: 639-672. [ Links ]
20. Weber MA, Giles TD. Inhibiting renin-angiotensin system to prevent cardiovascular deseases: do we need a comprehensive strategy?. Rev Cardiovasc Med 2006; 7: 45-54. [ Links ]
21. Gradman AH, Pinto R, Kad R. Current concepts: rennin inhibition in the treatment of hypertension. Curr Opin Pharmacol 2008; 8: 120-126. [ Links ]
22. World Health Organization. Global health risk. Mortality and burden of disease attributable to selected major risk. Geneva: World Health Organization,2009. [ Links ]
23. Manrique C, Lastra G, Gardner M, Sowers J. The renin angiotensin aldosterone system in hypertension: roles of insulin resistance and oxidative stress. Med Clin Am 2009;93:569-582. [ Links ]
24. Klein S, Fontana L, Young V, Coggan A, Kilo C, Patterson B, Mohammed B. Absence of an effect of liposuction on insulin action and risk factor for coronary heart disease. N Engl J Med 2004;350:2549-2557. [ Links ]
25. Sarzani R, Salvi F, Dessi P, Rappelli A. Renin-angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens 2008;26:831-843. [ Links ]
26. Aneja A, El-Atat F, McFarlane S, Sowers J. Hypertension and obesity. Recent Progr Horm Res 2004;59:169-205. [ Links ]
27. Messerli F, Sundgaard K, Reisin E, Dreslinski G, Ventura H, Oigman W, Frohlich E, Dunn F. Dimorphic cardiac adaptation to obesity and arterial hypertension. Ann intern Med 1983;99:757-761. [ Links ]
28. Harp J, Henry S, Di Girolamo M. Dietary weight loss decreases serum angiotensin-converting enzyme activity in obese adult. Obes Res 2002;10: 985-990. [ Links ]
29. Grassi G, DellOro R, Quarti F, Scopelliti F, Seravalle G, Paleari F, Gamba P, Mancia G. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia 2005;48: 1359-1365. [ Links ]
30. Rosa FJ, Lima Martínez M, Romero Vecchione E. Regulación Neuroendocrina de la función cardiovascular en el síndrome metabólico y diabetes. Interrelación con la sensibilidad a la sal. En: Soltero I editor. Aterosclerosis al Día VII. Caracas. Editado por Asociación Venezolana de Aterosclerosis AVA. 2009. p. 226-255. [ Links ]
31. Cooper S, Whaley A, Habibi J, Wei Y, Lastra G, Manrique C, Stas S, Sowers J. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol 2007;293:H2009-H2023. [ Links ]
32. Potter L, Abbey S, Dickey D. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev 2006; 27: 47–72. [ Links ]
33. Sarzani R, Dessi P, Paci M, Espinosa E, Rappelli A. Expression of natriuretic peptide receptors in human adipose and other tissues. J Endocrinol Invest 1996; 19:581–585. [ Links ]
34. Mehra M, Uber P, Park M, Scott R, Ventura H, Harris B, Frohlich E. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J Am Coll Cardiol 2004; 43:1590–1595. [ Links ]
35. Dessi P, Sarzani R, Rappelli A. The natriuretic peptide system in obesity-related hypertension: new pathophysiological aspects. J Nephrol 1998;11: 296–299. [ Links ]
36. Marrero MB, Fulton D, Stepp D, Stern DM. Angiotensin II-induced insulin resistance and protein tyrosine phosphatases. Arterioscler Thromb Vasc Biol 2004; 24: 2009-2013. [ Links ]
37. Lima M, Rosa F, Marin A. Síndrome metabólico y adiponectina. Informed 2008; 10: 195-201. [ Links ]
38. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 2005; 146: 1473-1481. [ Links ]
39. Olivares-Reyes JA, Shah BH, Hernandez-Aranda J, GarcIa-Caballero A, Farshori MP, Garcia-Sainz JA, Catt KJ. Agonist-induced interactions between angiotensin AT1 and epidermal growth factor receptors. Mol Pharmacol 2005; 68: 356-364. [ Links ]
40. Arellano-Plancarte A, Hernandez-Aranda J, Catt KJ, Olivares-Reyes JA. Angiotensin-induced EGF receptor transactivation inhibits insulin signaling in C9 hepatic cells. Biochem Pharmacol 2010; 79: 733-745. [ Links ]
41. Jansson L. The regulation of pancreatic islet blood flow. Diab Metab Rev 1994; 10: 407–416. [ Links ]
42. Carlsson PO. Angiotensin II and the endocrine pancreas: effects on islet blood flow and insulin secretion in rats. Diabetologia 1998; 41: 127–133. [ Links ]
43. Olivares-Reyes JA, Arellano A, Castillo JR. Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol 2009; 302: 128–139. [ Links ]
44. Sowers JR. Hypertension, angiotensin II and oxidative stress. N Engl J Med 2002; 346: 1999– 2001. [ Links ]
45. Matsuoka T. Glycation-dependent reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 1997; 99: 144–150. [ Links ]
46. Jaikaran ET, Clark A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochem Biophys Acta 2001; 1537: 179–203. [ Links ]
47. Niskanen L, Hedner T, Hansson L, for the CAPPP Study Group. Reduced cardiovascular morbidity and mortality in hypertensive diabetic patients on first-line therapy with an ACE inhibitor compared with a diuretic/ betablocker-based treatment regimen: a subanalysis of the Captopril Prevention Project. Diabetes Care. 2001; 24:2091–2096. [ Links ]
48. Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 2001, 37: 1053–1059. [ Links ]
49. Braga M, Leiter L. Role of renin-angiotensin system blockade in patients with diabetes mellitus. Am J Cardiol 2009; 104: 835-839. [ Links ]
50. Califf R, Boolell M, Haffber S, Bethel M, McMurray J, Duggal A, Holman R. Prevention of diabetes and cardiovascular disease in patients with impaired glucose tolerance: rationale and design of the nateglinide and valsartan in impaired glucose tolerance outcome research (NAVIGATOR) trial. Am Heart J 2008; 156: 623-632. [ Links ]
51. Oudit G, Liu GC, Zhong JC, Basu R, Chow FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM, Herzenberg AM, Kassiri Z, Scholey JW. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes 2010; 59: 529-538. [ Links ]
 Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
Todo el contenido de esta revista, excepto dónde está identificado, está bajo una Licencia Creative Commons
