










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. v.9 n.3 Mérida dic. 2011
La otra cara de Janus y la enfermedad tiroidea autoinmune
Joselyn Rojas 1,3, Miguel Angel Aguirre2,3, Raquel Cano3,4, Marjorie Villalobos2,3, Lisbeth Berrueta1
1Instituto de Inmunología Clínica – Universidad de los Andes. Mérida, Venezuela. 2Unidad de Endocrinología – Instituto Autónomo Hospital Universitario de los Andes. Mérida, Venezuela. 3Centro de Investigaciones Endocrino-Metabólicas "Dr. Félix Gómez – Universidad del Zulia. Maracaibo, Venezuela.4 Unidad de Endocrinología y Enfermedades Metabólicas del Hospital Universitario de Caracas. Caracas – Venezuela.
Dirigir correspondencia a: Dra. Joselyn Rojas. E-mail: rojas.joselyn@gmail.com.
RESUMEN
La enfermedad tiroidea autoinmune es una de las patologías más comunes dentro de la endocrinología. Este grupo de enfermedades que afectan la función tiroidea se consideran actualmente como parte de un espectro, donde por un lado se evidencia una hipofunción y donde predomina una respuesta inmune Th1 (Tiroiditis de Hashimoto) y en el lado opuesto, el de la hiperfunción, predomina una respuesta Th2 (Enfermedad de Graves). Si bien ambas enfermedades tienen ciertos locus de predisposición genética en común, las manifestaciones clínicas y el pronóstico de estas enfermedades son totalmente opuestas. Aunque se han planteado diversas teorías para explicar las diferencias en el patrón autoinmune de ambos extremos de este espectro, éstas no han logrado ilustrar la amplia variedad de fenotipos de esta patología. Evidencia actual sugiere que un grupo celular encargado de controlar la actividad de los elementos efectores de la respuesta inmune, limitando el daño hacia los tejidos del hospedador mediando la autotolerancia, y conocido como células T reguladoras, juegan un papel protagónico en el desarrollo de las enfermedades autoinmunes. El propósito de ésta revisión es exponer como la disregulación de la cara reguladora del sistema inmune, expresada tanto en la alteración de la función como en la expansión de las células T reguladoras, constituyen una piedra angular en las diversas formas de manifestación de la enfermedad tiroidea autoinmune.
Palabras clave: enfermedad tiroidea autoinmune, células T reguladoras, enfermedad de Graves, tiroiditis de Hashimoto.
ABSTRACT
Autoimmune thyroid disease is one of the most common diseases inside the field of endocrinology. This group of diseases affect the thyroid function in a manner that is now considered like a spectrum, where on one side there is evidence of hypofunction and a predominating Th1 response (Hashimoto´s thyroiditis), and on the opposite side, hyperfunction and a predominating Th2 response (Graves´ disease). Even though both diseases share loci for genetic predisposition, the outcome of each of them is totally opposite. Several theories have been proposed to explain the differences in the autoimmune patterns, but havent been able to explain the ample phenotypical manifestations of the disease. Current evidence suggests that a recently describe cellular group is in charge of controlling the effector elements of the immune response, limiting tissue damage and enhancing self-tolerance, these are the T regulator cells (Treg), which play a principal role in the development of autoimmune diseases. The purpose of this review is to expose the dysregulation of the regulatory side of the immune system expressed as the alteration of the functioning of Treg, which are now believed to be fundamental in the many phenotypes of autoimmune thyroid diseases.
Key words: autoinmune thyroid disease, T regulator cell, Graves´ disease, Hashimoto´s thyroiditis.
Artículo recibido en: Agosto 2011. Aceptado para publicación en: Septiembre 2011.
Para la mitología Romana, el Dios Janus fue una deidad capaz de ver hacia el pasado y hacia el futuro (aunque de forma académica se denomina "el dios de los principios y las transiciones"), siendo simbolizado con dos caras que miraban al este y al oeste respectivamente1. De acuerdo con los escritos, él dominaba los Cielos y todo aquel que quisiera entrar debía pasar por él, inclusive el mismo Júpiter, por lo que se postula como el dios más importante de la mitología. Debido a su capacidad de observar ampliamente como inician y finalizan diferentes procesos (en especial los relacionados con agricultura), Janus es el dios de las puertas y entradas, siendo venerado en los solsticios, inicio de cosechas, inclusive se ha llegado a considerar como el creador del Hombre ya que es la deidad que preside el comienzo de cualquier cosa u evento1.
Al extrapolar este escenario hacia el contexto fisiológico de la respuesta inmune, este sistema inmune actuaría como una dualidad efectora y reguladora, en la cual existe un equilibrio entre elementos efectores y los que regulan dicha actividad. Una de las caras del sistema inmune está compuesta por subtipos celulares capaces de ejercer la función de defensa, abarcando tanto células del sistema innato como del adaptativo, por ejemplo las células asesinas naturales o "natural killer" (NK), monocitos/macrófagos, polimorfonucleares, linfocitos B, linfocitos T CD4+ y CD8+; mientras que su contraparte abarca todas las poblaciones celulares encargadas de regular la duración, extensión y cese de las respuestas efectoras. En esta revisión se analizará la otra cara de Janus, el papel de las células reguladoras en los fenómenos de autoinmunidad asociados a la enfermedad tiroidea autoinmune (ETAI).
CONCEPTOS BÁSICOS
Se considera autoinmunidad al fenómeno en el cual el sistema inmune efector se vuelca hacia las células propias (sanas), o dicho en otras palabras, cuando existe una respuesta inmune hacia antígenos del hospedador (auto-antígenos)2.
La patogenia de la autoinmunidad es mucho más compleja de lo que se proponía en el pasado, involucrando factores moleculares, ambientales y genéticos en el desarrollo y progresión de dichas enfermedades, como por ejemplo: mimetismo molecular, activación por espectador inocente, polimorfismos del antígeno leucocitario humano (HLA), modificaciones post-traduccionales de proteínas, mutaciones puntuales de factores de transcripción fundamentales, y pérdida de la tolerancia central y/o periférica. Desde un punto de vista evolutivo, el sistema inmune adaptativo aparece en la llamada Catástrofe Oxigénica3 o el Big Bang Evolucionario4 hace 600 millones de años. Antes de la aparición de los vertebrados con mandíbula (gnathostomos), los seres vivos desarrollaron un sistema de defensa "primitivo" innato, basado en el desarrollo de receptores genéricos los cuales reconocían patrones moleculares conservados y eran capaces de activar una respuesta inflamatoria4. Al aparecer en los gnathotosmos, se inicia la evolución de un sistema de defensa más avanzado en el cual la memoria inmunológica es fundamental para el reconocimiento de patógenos en encuentros posteriores, mediando una respuesta efectiva de defensa, basada en elementos tanto humorales (anticuerpos) como celulares4. Si bien, ambos sistemas de defensa se han mantenido en los vertebrados superiores, la clave de su preservación ha sido el poder defender al hospedador sin generar daño en los tejidos propios. Las células reguladoras forman parte del grupo celular encargado de controlar la actividad de los elementos efectores de la respuesta, limitando el daño hacia los tejidos del hospedador, mediando la auto-tolerancia.
Existen varios fenotipos de células reguladoras según el origen: células T reguladoras (Treg) naturales CD4+CD25+Foxp3+ (nTreg), células T reguladoras inducibles CD4+ (iTreg: Tr1 y Th3), células T reguladoras CD8+CD25+ y CD8+CD28-, las células NKT y las células T γδ; (ver Figura 1).

nTreg
Desde la descripción de Sakaguchi y col.5 acerca del papel del factor de transcripción Foxp3 en la generación de las células T reguladoras, una ola de investigación y hallazgos concernientes al desarrollo, inmunofenotipos y función de las Treg ha inundado la comunidad científica. El subgrupo de Treg más común es el CD4+CD25+Foxp3+GITR+CTLA-4+CD62L+CD127+OX40L+TNFR2+TGFBR1+LAG3+, capacitado para mediar control sobre la célula blanco, mediante mecanismos célula-célula: TGF-β1 anclado en la membrana de la Treg, vía granzyma/perforina, y mediado por LAG-36-10. Otros fenotipos de nTreg incluyen: CD4lowCD25+Foxp3+LAG3low (los cuales presentan alta granularidad) y los CD4+ Foxp3+Gadd45α/β+.
iTreg
Las células reguladoras inducidas en la periferia dependen del estímulo mediado por citocinas producidas en el nódulo linfático en presencia de actividad antigénica. El primer subtipo, las Tr1 se originan cuando una célula CD4+ naive es estimulada por IL-10, generando una célula CD4+CD25lowFoxp3lowIL10hi, las cuales producen muy poco TGF-β1 e IFN-γ y son capaces de mediar acciones citotóxicas o dependientes de citocinas sobre sus células blanco6-10. Las Th3 aparecen cuando el estímulo sobre la CD4+ naive es el TGF-β1 e IL-4, originando una CD4+CTLA-4+TGFβ+, capaz de modular actividad celular mediado por citocinas6-10. Otros inmunofenotipos de iTreg incluyen: CD4+CD25+CTLA-4+Foxp3lowGITRlow (inducidas mediante la acción de células dendríticas plasmocitoides sobre CD4+ naive), y CD4+CD25+CTLA-4+Foxp3+GITR+ (originadas de la inducción de CD4+CD25low con TGF-β1).
CD8+ REGULADORAS
Consideradas como las células citotóxicas por excelencia, junto a las NK, su papel como células con potencial regulador se ha reconocido desde hace más de 15 años. Al igual que sus homólogos CD4+, las CD8+ reguladoras pueden ser naturales o inducidas, con un inmunofenotipo común de CD8+CD25+CD28-CTLA-4+GITR+Foxp3+, las cuales suprimen la respuesta inmune mediante contacto célula-célula o dependiente de citocinas como IL-10/TGF-β111-15. Otro mecanismo de inducción de CD8+CD25+Foxp3+ es mediante estimulación antigénica continua de monocitos CD14+ 16. Otros subtipos menores incluyen: CD8+CD122+ con características de célula de memoria, y CD8+CXCR3+IL-10+ con actividad supresora de la producción de IFN-γ. Dentro del grupo de las iTreg CD8+ tenemos los CD8+CD103+ que son inducidos por aloestimulación durante reacciones injerto vs huésped en presencia de IL-10, IL-4 y TGF-β17, y los CD8+CD103+Foxp3+GITR+CTLA-4+ inducidos por péptidos virales provenientes de virus de la hepatitis C (HCV) y virus de la gripe18.
NKT
Las células T "Natural Killer" (NKT) conforman un grupo de linfocitos T, los cuales expresan el receptor αβ de célula T que reconoce antígenos lipídicos presentados por el complejo mayor de histocompatibilidad (MHC) clase I CD1d, y marcadores de superficie de NK como el Nk1.119-21. El subtipo de NKT- I (iNKT) se caracteriza por la expresión de la cadena α invariante del TCR (Vα24-Jα18) en conjunción con la cadena β Vβ11, y su ligando prototipo es la α-galactosil-ceramida (presentado por CD1d); el subtipo NKT- II, presenta un amplio repertorio αβTCR, los cuales son capaces de reconocer antígenos lipídicos como los sulfatides pero no reconocen α-galactosil-ceramida. El inmunofenotipo de las iNKT incluye CD62L-CD69+CD44hiIL2RhiCCR2+CCR5+CXCR2+CXCR3+CXCR4+, algunos CD4+ (CCR4+) o doble negativos (CCR1+CCR6+CCL3+CCL20+ [DN]). De acuerdo al gradiente citoquímico presente durante las diferentes fases de la respuesta inflamatoria, ocurrirá la migración de las iNKT CD4+ o DN, funcionando como moduladores de la respuesta de células T helper (secreción de IFN-γ, IL-4, IL-5, IL-13 y IL-17) o mediando destrucción de células blanco (células dendríticas o células T efectoras) mediante la vía perforina/granzyma.
CÉLULAS T γδ
Este subtipo de célula T expresa un receptor TCR con cadenas γδ, en vez de las canónicas αβ observadas en el resto de las subpoblaciones, las cuales además se consideran nulas para CD4 y CD822-24. Los antígenos reconocidos por este grupo celular pueden ser péptidos o no peptídicos (alquilamidas y pirofosfatos) presentados por MHC de clase I (MIC-A, -B y CD1d [restringidos como las iNKT]), los cuales pueden incluso no requerir procesamiento por células presentadoras de antígenos. Una vez activadas, son productoras masivas de TNF-α e IFN-γ, induciendo la polarización hacia respuesta tipo Th1.
CONTROL DE LA RESPUESTA INMUNE
Como se describió previamente, los diferentes subtipos de células reguladoras poseen mecanismos particulares de control de la actividad de la célula blanco, ya sea mediante inducción de indolamina 2,3-dioxigenasa (IDO), muerte celular, manipulación de la maduración de la célula dendrítica o por influencia citoquímica (ver Figura 2). La importancia de estos mecanismos sobre el control de fenómenos de autoinmunidad será explicada a continuación analizando la patogenia de la ETAI.
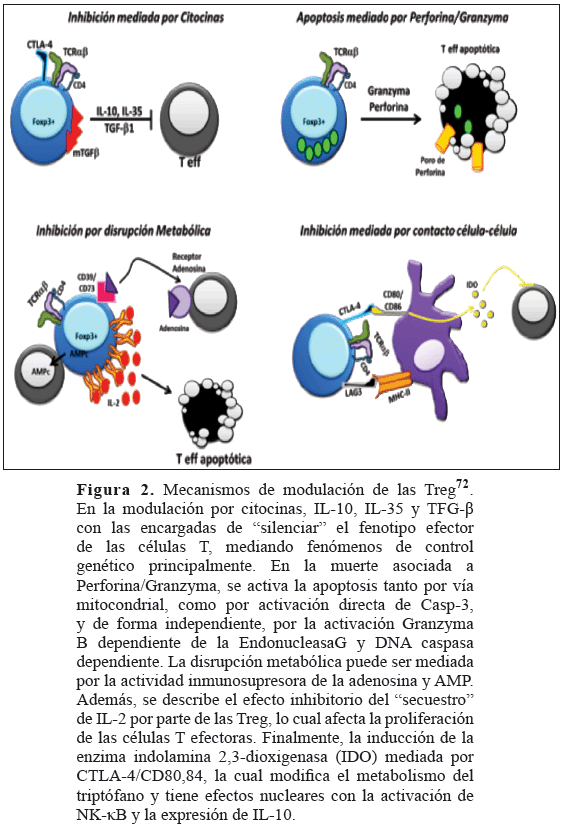
ENFERMEDAD TIROIDEA AUTOINMUNE (ETAI)
Este grupo de enfermedades que afectan la función tiroidea se consideran actualmente como parte de un espectro, donde por un lado predomina una respuesta Th1 (Tiroiditis de Hashimoto) y en el lado opuesto predomina una respuesta Th2 (Enfermedad de Graves). Si bien ambas enfermedades (podría decirse) tienen ciertos locus de predisposición genética comunes (polimorfismos de HLA: DRB1*03 para Graves, y HLA-DR3, HLA-DR4 y HLA-DQw7DQB1*0301 para Hashimoto)25, polimorfismos de CTLA-4, AIRE, PTPN22, tiroglobulina (Tg), peroxidasa tiroidea (TPO) y receptor de TSH26-28, inclusive polimorfismos de Foxp329, las manifestaciones clínicas y el pronóstico de estas enfermedades es totalmente opuesto. Se han planteado varias hipótesis para explicar esta variación, tomando en consideración diversos factores ambientales como la ingesta de yodo30, polimorfismos del transportador Na+/I- 31, y otros, pero no ha sido suficiente para explicar la amplia variedad de manifestaciones en el espectro. De acuerdo a la información actual, uno de los mecanismos propuestos es la pérdida de regulación de las células Treg.
Marazuela y col.32 realizaron un ensayo clínico en el cual caracterizaron las Treg CD4+ de pacientes con ETAI, tanto en sangre como en biopsias tiroideas. Este grupo reporta que los niveles de CD4+GITR+, CD4+Foxp3+, CD4+ sintetizadoras de IL-10 o TGF-β fueron significativamente mayores en pacientes con ETAI en comparación con sujetos normales; sin embargo, no se evidenció diferencia en los niveles de dichos subtipos celulares entre pacientes con tiroiditis de Hashimoto (TH) y aquellos con enfermedad de Graves (EG); estos hallazgos son concordantes con otro ensayo en pacientes con EG donde tampoco se encontró déficit de Tregs33. Al enfrentar las Treg con anticuerpos neutralizantes in vitro, se observó que las Treg presentaban defectos al momento de ejercer su función supresora, probablemente debido a refractariedad de las células efectoras blanco o por incremento en la expresión de GITRL34, el cual se ha reconocido como capaz de abolir la actividad supresora de las Treg, incrementando a su vez su vida media.
Al año siguiente, McLachlan y col.35 publican sus resultados utilizando un modelo animal que permitía evaluar la progresión de EG hacia TH analizando la expansión de los epítopes antigénicos durante la evolución de la enfermedad. Utilizando un modelo transgénico que expresa la subunidad-A del receptor de TSH humano (TSHrA), evidenciaron que la progresión de EG hacia TH depende del número y función de las Treg, especialmente manipulando el cambio de epítope hacia aquellos crípticos como TPO y Tg. La depleción experimental de CD4+CD25+ generó tiroiditis moderada incluso antes de la inmunización con TSHr/Subunidad-A como antígeno, y posterior a la inmunización se obtuvo una tiroiditis
severa (~80% de la glándula) que progresó a hipotiroidismo franco. Una vez desencadenada la destrucción tiroidea, aparecieron autoanticuerpos para TPO y Tg, lo cual concuerda con la progresión observada en algunos casos humanos. A pesar de comprobar que las Treg modulan la protección contra la aparición de nuevos epítopes propios y controla la progresión de la autoinmunidad tiroidea, en este estudio no se pudo concluir cuál era la causa del defecto en este grupo celular.
Los trabajos anteriormente mencionados (entre otros36-37), plantean que las Treg son fundamentales en la patogenia de autoinmunidad tiroidea, caracterizándose por defectos en su función supresora pudiendo estar acompañado de disminución en su número38. Ahora bien, que significa que una célula Treg sea defectuosa? Kriegel y col.39 evaluaron estas características en células obtenidas de pacientes con síndrome poliglandular autoinmune (SPA) tipo II, demostrando que las Treg de dichos pacientes tenían un buen número de células, con marcadores de superficie que indicaban madurez, pero eran incapaces de modular la proliferación celular, datos que son similares a los del grupo de Marazuela. Sin embargo, el grupo Alemán no encontró incremento en GITR39. Varios han avalado el papel de éste receptor en la inhibición de la actividad de las Treg40-42, por lo que es tentador plantear que los defectos observados en ETAI puedan deberse a activación continua del receptor por ligandos sintetizados en las células presentadoras de antígeno (APC) locales, y por el tiroidocito, especialmente cuando el mismo receptor es activador de las células efectoras, con liberación de citocinas Th1, lo que parcialmente explicaría la progresión del daño tisular tiroideo. De hecho, modelos animales GITR-/- 43-45 o tratados con anticuerpos monoclonales anti-GITR46-47 están protegidos contra fenómenos de autoinmunidad, asociado a la falta de inducción de una respuesta Th1, con implicaciones incluso como terapia adyuvante oncológica. A pesar de la evidencia antes descrita, van Olffen y col.48 reportan que GITR ligando es un inductor directo de CD4+ reguladoras, pero no CD8+ reguladoras, siendo este efecto mediado por la producción de IL-2; por ende, los autores finalizan proponiendo que GITR ligando es factor de proliferación de Treg.Otro candidato a la modulación de la actividad de Treg, es el ligando inductor de apoptosis relacionado con TNF (TRAIL), el cual ha acumulado evidencia controversial suficiente sobre su papel en autoinmunidad. En los últimos 15 años, experimentos en modelos animales para diabetes tipo 149, artritis reumatoidea50 y tiroiditis autoinmune experimental51 han demostrado que el silenciamiento genético del TRAIL y su aplicación exógena en fases preclínicas y clínicas de enfermedad autoinmune, controlan y son capaces de revertir la progresión de la misma, a través de la inducción de CD4+CD25+CD45RBlow productoras de IL-10, TGF-β1, con capacidad supresora conservada, llegándose a proponer un mecanismo de control antiinflamatorio entre células epiteliales tiroideas y células inflamatorias52. Sin embargo, Ikeda y col.53 publican el año pasado que los efectos de TRAIL sobre Treg y células tiroideas dependen del tipo de receptor estimulado y la presencia de receptores tipo "decoy" o señuelo (receptor de evasión). TRAIL tiene dos receptores naturales, receptor de muerte 4 y 5 (DR4 y DR5), y 3 receptores decoy, DcR1 (no tiene dominio citoplasmático), DcR2 (dominio de muerte truncado) y DcR3 (actividad supresora de vías de FasL). Los datos de Ikeda proponen entonces que según el juego de concentraciones de receptores reales y sus decoy podrían modular la actividad de TRAIL sobre Treg, lo cual explica los hallazgos de Xiao y col.54 en la cual se observó incremento en la expresión y secreción de TRAIL en células T efectoras, acompañado de apoptosis de Treg a partir de muestras de pacientes con artritis reumatoidea. La apoptosis inducida por TRAIL no es exclusiva para las Treg. Se ha demostrado muerte celular en células foliculares tiroideas mediado por TRAIL/DR5 en presencia de exceso yodo en modelos de tiroiditis autoinmune experimental55. Finalmente, IFN-γ y TNF-α son capaces de inducir la expresión del receptor DR5 en células epiteliales tiroideas, lo cual la hace sensibles a los efectos apoptóticos de TRAIL56.
Existen otras posibilidades a ser tomadas en cuenta con respecto al defecto supresor observado en la ETAI, tal es el caso de la esfingosina-1-fosfato (S1P), PKCθ, y la disfunción de las APC. La S1P, metabolito esfingolípido activo producto de la esfingosina kinasa, es una molécula de señalización intracelular asociada con metabolismo de calcio intracelular, degranulación de polimorfonucleares y mastocitos, movimiento celular, inhibidor de la apoptosis, e involucrada en la termotolerancia57-59. Para el sistema inmune, la S1P es la molécula fundamental para la salida del timo y homing adecuado de los timocitos60. Utilizando enfoques elegantes de adquisición y pérdida de función de genes, Liu y col.61 han reportado que S1P, a través de su receptor acoplado a proteína G S1P1, es capaz de suprimir la diferenciación y actividad supresora de Treg, a través de la vía Akt-mTOR, lo cual ofrece un nuevo panorama a las posibilidades de intervención a través de la modulación de esta vía. La proteína kinasa θ es punto clave en la cascada de activación de las células T, vía TCR 62, aunque su papel en la proliferación y actividad de las Treg es controversial. Gupta y col.63 publicaron en 2009 que la PKCθ es esencial para la proliferación y diferenciación de Treg, mediante la inducción de Foxp3 vía calcineurina/NFAT. Zanin-Zhorov y col.64 publican que la enzima es secuestrada lejos del área de la sinapsis inmunológica durante la activación de las Treg, y el bloqueo de la misma en modelos in vitro incrementa la actividad de las Treg, sugiriendo que durante la inducción de la actividad supresora ésta enzima actúa de forma antagónica. Se propone que la modulación de PKCθ, es un posible blanco terapéutico en autoinmunidad, especialmente porque es uno de los mediadores cascada abajo del TNF-α65. Por último, se encuentra la disfunción de las APC la cual fue correlacionada con el grado de actividad supresora de las Treg en pacientes con diabetes tipo 166. Este grupo observó que si bien, en autoinmunidad se observan defectos en la activación de Treg, parte del problema se localiza en la actividad de las APC a la hora de inducirlas, originando una muy baja actividad supresora por parte de Treg (menos del 25%) en ~40% de los pacientes analizados.
PERSPECTIVAS
No cabe duda que las células reguladoras son un punto clave en la patogenia de las enfermedades autoinmunes, y en el caso presentado en esta revisión, el problema no es solamente de expansión, también deben analizarse desde un punto de vista funcional. Las Treg son quizá las células más importantes en el grupo, pero eso no significa que sean las únicas alteradas en la ETAI. Por ejemplo, recientemente se publicó que iNKT pueden autopresentar Tg sin necesidad de presentación antigénica por parte de una APC, asociado a la producción exagerada de IFN-γ y/o TNF-α, lo que polariza la respuesta hacia Th1 en modelo animal de tiroiditis autoinmune inducido por yodo67. Las células T γδ están relacionadas con el balance Th1/Th17 lo cual es fundamental para la progresión de la tiroiditis experimental. Las CD4+ γδ bajo la influencia de IL-1β e IL-23 pueden secretar IL-17, IL-21 e IL-22, lo cual polariza el desarrollo de Tnaive hacia Th17, células patogénicas en autoinmunidad68 y especialmente en la progresión de la TH69 y en la refractariedad de la EG70. Finalmente, la frecuencia de poblaciones CD8+CD25+ junto a la aparición progresiva de autoanticuerpos, se correlacionan con la severidad de la TH71. Con toda esta información, no queda más que plantear un esquema de eventos que se suscitan en el espectro de la ETAI y que permiten dar entendimiento a esta complicada enfermedad (Figura 3).

A pesar de la vasta información existente y de las evidencias experimentales reportadas, aún quedan muchas interrogantes por responder: ¿Por qué los autoanticuerpos para TPO aparecen después de Tg? ¿Por qué hay pacientes positivos para ellos sin evidencia de destrucción glandular severa? ¿Qué impide el daño tisular en los cuadros de hipertiroidismo o hipotiroidismo subclínicos con autoanticuerpos positivos y glándula preservada? ¿Cuáles son las conexiones fisiopatológicas precisas entre ETAI y diabetes tipo 1, y con ellos el resto de los SPA?
Uno de los retos más importantes es comprender el sistema de control apoptótico entre las Treg, las T efectoras y los tiroidocitos. Es menester evaluar la expresión de los DR4, DR5 y los decoy en biopsias de pacientes con TH y EG, ya que eso permitirá esclarecer el patrón de muerte según la polarización de Th en la cual se encuentre el paciente. Como se muestra en la Figura 3, se propone que existe una modulación en la expresión y localización de DR4/5 y decoys tanto en Treg como en tiroidocitos lo que pudiera explicar la variabilidad clínica en la ETAI; teoría que concuerda con lo observado en líneas cancerosas resistentes a apoptosis, en las cuales se ha observado que el tratamiento con TRAIL modifica la distribución de los receptores en las balsas lipídicas y altera su sensibilidad a la apoptosis mediada por el ligando72. Además, sería interesante evaluar la capacidad de TRAIL de disminuir la expresión de cFLIP en tiroidocitos, dando respuestas al perfil apoptótico antagónico entre TH y EG73. Por último, evaluar la actividad de S1P dentro del espectro de la inmunopatogenia, a la luz de su efecto sobre las Treg, debido a que ya existe evidencia de su papel oncogénico en el cáncer tiroideo74 asociándose a un fenotipo pro-migratorio (metastásico) dependiente de PKCα y ERK1/2, y su influencia en otras enfermedades autoinmunes como el Síndrome de Sjögren75.
Todos los sistemas fisiológicos tienen una doble cara: en exceso y en ausencia. Para el caso de la ETAI es probable que además de la herencia y los factores ambientales, exista un funcionamiento defectuoso de las células reguladoras, lo que para el caso de TH eterniza la polarización hacia Th1, mientras que en EG mantenga una polarización Th2. Utilizar la analogía del dios Janus para explicar duplicidad en eventos de una misma vía metabólica, etiología, o manifestación clínica, es un buen ejemplo para demostrar que no hay absolutos, todo depende hacia donde se desplace la balanza de la transición.
REFERENCIAS BIBLIOGRÁFICAS
1. Hardie P. The Janus Episode in Ovid´s Fasti. Materiali e discussioni per l´analisis dei testi classici 1991;26:47-64. [ Links ]
2. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky´s postulates revisited). Immunol Today 1993;14:426-430. [ Links ]
3. Dismukes GC, Klimov VV, Baranov SV, Kozlov YN, DasGupta J, Tyryshkin A. The origin of atmospheric oxygen on earth: the innovation of oxygenic photosynthesis. Proc Natl Acad Sci U S A 2001;98:2170-2175. [ Links ]
4. Cooper MD, Alder MN. The evolution of adaptative immune systems. Cell 2006;124:815-822. [ Links ]
5. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by transcription factor Foxp3. Science 2003;299:1057-1061. [ Links ]
6. Bluestone JA, Abbas AK. Natural versus adaptative regulatory T cells. Nat Rev Immunol 2003;3:253-257. [ Links ]
7. Fehérvari Z, Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest 2004;114:1209-1217. [ Links ]
8. Lan RY, Ansari AA, Lian ZX, Gershwin ME. Regulatory T cells: development, function and role in autoimmunity. Autoimmun Rev 2005;4:351-363. [ Links ]
9. La Cava A. Tregs are regulated by cytokines: implications for autoimmunity. Autoimmun Rev 2008;8:83-87. [ Links ]
10. Beyer M, Schultze JL. Plasticity of Tregs cells: is reprogramming of Treg cells possible in the presence of FOXP3?. Int Immunopharmacol 2011;11:555-560. [ Links ]
11. Suzuki M, Konya C, Goronzy JJ, Weyand CM. Inhibitory CD8+ T cells in Autoimmunity disease. Hum Immunol 2008;69:781-789. [ Links ]
12. Smith TR, Kumar V. Revival of CD8+ Treg-mediated suppression. Trends Immunol 2008;29:337-342. [ Links ]
13. Lu L, Cantor H. Generation and regulation of CD8+ regulatory T cells. Cell Mol Immunol 2008;5:401-406. [ Links ]
14. Vinay DS, Kwon BS. CD11c+CD8+ T cells: two-faced adaptative regulators. Cell Immunol 2010;264:18-22. [ Links ]
15. Pomié C, Vicente R, Vuddamalay Y, Lundgren BA, van der Hoek M, Enault G, Kagan J, Fazilleau N, Scott HS, Romagnoli P, van Meerwijk JPM. Autoimmune regulator (AIRE)-deficient CD8+CD28low regulatory T lymphocytes fail to control experimental colitis. Proc Natl Acad Sci U S A 2011;108:12437-12442. [ Links ]
16. Mahic M, Henjum K, Yaqub S, Bjornbeth BA, Togersen KM, Tasken K, Aandahl EM. Generation of highly suppressive adaptative CD8(+)CD25(+)FOXP3(+) regulatory T cells by continuous antigen stimulation. Eur J Immunol 2008;38:640-646. [ Links ]
17. Uss E, Rowshani AT, Hooibrink B, Lardy NM, van Lier RA, ten Berge IJ. CD103 is a marker for alloantigen-induced regulatory CD8+ T cells. J Immunol 2006;177:2775-2783. [ Links ]
18. Billerbeck E, Blum HE, Thimme R. Parallel expansion of human virus-specific Foxp3-effector memory and de novo-generated Foxp3+ regulatory CD8+ Y cells upon antigen recognition in vitro. J Immunol 2007;179:1039-1048. [ Links ]
19. Gapin L. iNKT cell autoreactivity: what is "self" and how it is recognized?. Nat Rev Immunol 2010;10:272-277. [ Links ]
20. Taniguchi M, Tashiro T, Dashtsoodol N, Hongo N, Watari H. The specialized iNKT cell system recognizes glycolipids antigens and bridges the innate and acquired immune systems with potential applications for cancer. Int Immunol 2009;22:1-6. [ Links ]
21. Reilly EC, Wands JR, Brossay L. Cytokine dependent and independent iNKT cell activation. Cytokine 2010;51:227-231. [ Links ]
22. Vallejo AN, Davila E, Weyand CM, Goronzy JJ. Biology of T lymphocytes. Rheum Dis Clin N Am 2004;30:135-157. [ Links ]
23. Spicuglia S, Bonnet M, Ferrier P. Alpha/beta versus gamma/delta T cell development: a choice linked to the transcription factor Sox13. Med Sci (Paris) 2007;23:457-458. [ Links ]
24. Born WK, Zhang L, Nakayama M, Jin N, Chain Huang Y, Aydintug MK, O´Brien RL. Peptide antigens for gamma/delta T cells. Cell Mol Life Sci 2011;68:2335-2343. [ Links ]
25. Jacobson EM, Huber A, Tomer Y. The HLA gene complex in thyroid autoimmunity: from epidemiology to etiology. J Autoimmun 2008;30:58-62. [ Links ]
26. Ban Y, Tomer Y. Susceptibility genes in thyroid autoimmunity. Clin Dev Immunol 2005;12:47-58. [ Links ]
27. Jacobson EM, Tomer Y. The CD40, CTLA-4, thyroglobulin, TSH receptor and PTPN22 gene quintet and its contribution to thyroid autoimmunity: back to the future. J Autoimmun 2007;28:85-98. [ Links ]
28. Huber A, Menconi F., Corathers S, Jacobson EM, Tomer Y. Joint genetic susceptibility to type 1 diabetes and autoimmune thyroiditis: from epidemiology to mechanisms. Endocr Rev 2008;29:697-725. [ Links ]
29. Ban Y, Tozaki T, Tobe T, Ban Y, Jacobson EM, Concepcion ES, Tomer Y. The regulatory T cell gene FOXP3 and genetic susceptibility to thyroid autoimmunity: an association analysis in Caucasian and Japanese cohorts. J Autoimmun 2007;28:201-207. [ Links ]
30. Yamazaki K, Tanigawa K, Suzuki K, Yamada E, Yamada T, Takano K, Obara T, Sato K. Iodine-induced chemokines and genes related to immunological function in cultured human thyroid follicles in the presence of thyrotropin. Thyroid 2010;20:67-76. [ Links ]
31. Bizhanova A, Kopp P. The sodium-iodide symporter NIS and pendrin in iodide homeostasis of the thyroid. Endocrinology 2009;150:1084-90. [ Links ]
32. Marazuela M, García-López MA, Figueroa-Vega N, de la Fuente H, Alvarado-Sánchez B, Monsiváis-Urenda A, Sánchez-Madrid F, González-Amaro R. Regulatory T cells in human autoimmune thyroid disease. J Clin Endocrinol Metab 2006;91:3639-3646. [ Links ]
33. Pan D, Shin YH, Gopalakrishnan G, Hennessey J, De Groot LJ. Regulatory T cells in Graves´ disease. Clin Endocrinol 2009;71:587-593. [ Links ]
34. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 2002;3:135-142. [ Links ]
35. McLachlan SM, Nagatama Y, Pichurin PN, Mizutori Y, Chen CR, Misharin A, Aliesky HA, Rapoport B. The link between Graves´ disease and Hashimoto´s Thyroiditis: a role for regulatory T cells. Endocrinology 2007;148:5724-5733. [ Links ]
36. Morris GP, Brown NK, Kong YC. Naturally existing CD4+CD25+Foxp3+ regulatory T cells are required for tolerance to experimental autoimmune thyroiditis induced by either exogenous or endogenous autoantigen. J Autoimmun 2009;33:68-76. [ Links ]
37. Nagayama Y, Horie I, Saitoh O, Nakahara M, Abiru N. CD4+CD25+ naturally ocurring regulatory T cells and not lymphopenia play a role in the pathogenesis of iodide-induced autoimmune thyroiditis in NOD-H2h4 mice. J Autoimmun 2007;29:195-202. [ Links ]
38. Mao C, Wang S, Xiao Y, Xu J, Jiang Q, Jin M, Jiang X, Guo H, Ning G, Zhang Y. Impairment of regulatory capacity of CD4+CD25+ regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves´ disease. J Immunol 2011;186:4734-4743. [ Links ]
39. Kriegel MA, Lohmann T, Gabler C, Blank N, Kalden JR, Lorenz HM. Defective suppressor function of Human CD4+CD25+ Regulatory T cells in autoimmune polyglandular syndrome type II. J Exp Med 2004;199:1285-1291. [ Links ]
40. Burckhart T, Thiel M, Nishikawa H, Wüest T, Müller D, Zippelius A, Ritter G, Old L, Shiku H, Renner C. Tumor specific crosslinking of GITR as costimulation for immunotherapy. J Immunother 2010;33:925-934. [ Links ]
41. You S, Poulton L, Cobbold S, Liu C-P, Rosenzweig M, et al. Key role of the GITR/GITRLigand pathway in the development of murine autoimmune diabetes: a potential therapeutic target. PLoS One 2009;4:e7848. [ Links ]
42. Tomizawa R, Watanabe M, Inoue N, Takemura K, Hidaka Y, Akamizu T, Hayakawa K. Association of functional GITR gene polymorphisms related to expression of glucocorticoid-induced tumour necrosis factor-receptor molecules with prognosis of autoimmune thyroid disease. Clin Exp Immunol 2011;165:141-147. [ Links ]
43. Cuzzocrea S, Ayroldi E, Di PR, Agostini M, Mazzon E, Bruscoli S, Genovese T, Ronchetti S, Caputi AP, Riccardi C. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J 2005;19:1253-1265. [ Links ]
44. Santucci L, Agostini M, Bruscoli S, Mencarelli A, Ronchetti S, Ayroldi E, Morelli A, Baldoni M, Riccardi C. GITR modulates innate and adaptive mucosal immunity during the development of experimental colitis in mice. Gut 2007;56:52-60. [ Links ]
45. Nocentini G, Cuzzocrea S, Genovese T, Bianchini R, Mazzon E, Ronchetti S, Esposito E, Rosanna DP, Bramanti P, Riccardi C. Glucocorticoid-induced tumor necrosis factor receptor-related (GITR)-Fc fusion protein inhibits GITR triggering and protects from the inflammatory response after spinal cord injury. Mol Pharmacol 2008;73:1610-1621. [ Links ]
46. Piao J, Kamimura Y, Iwai H, Cao Y, Kikuchi K, et al. Enhancement of T-cell-mediated anti-tumour immunity via the ectopically expressed glucocorticoid-induced tumour necrosis factor receptor-related receptor ligand (GITRL) on tumours. Immunology 2009;127:489–499. [ Links ]
47. You S, Poulton L, Cobbold S, Liu C-P, Rosenzweig M, et al. Key Role of the GITR/GITRLigand Pathway in the Development of Murine Autoimmune Diabetes: A Potential Therapeutic Target. PLoS One 2009;4:e7848. [ Links ]
48. van Olffen RW, Koning N, van Gisbergen KP, Wensveen FM, Hoek RM, Boon L, Hamann J, van Lier RA, Nolte MA. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol 2009;182:7490-7500. [ Links ]
49. Mi QS, Ly D, Lamhamedi-Cherradi E, Salojin KV, Zhou L, Grattan M, Meagher C, Zucker P, Chen YH, Nagle J, Taub D, Delovitch TL. Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes 2003;52:1967-1975. [ Links ]
50. Yao Q, Wang S, Gambotto A, Glorioso JC, Evans CH, Robbins PD, Ghivizzani SC, Oligino TJ. Intra-articular adenoviral-mediated gene transfer of trail induces apoptosis of arthritic rabbit synovium. Gene Ther 2003;10:1055-1060. [ Links ]
51. Wang SH, Chen GH, Fan Y, Van Antwerp M, Baker JR Jr. Tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis by the expansion of CD4+CD25+ regulatory T cells. Endocrinology 2009;150:2000-2007. [ Links ]
52. Fang Y, Sharp GC, Yagita H, Braley-Mullen H. A critical role for TRAIL in resolution of granulomatous experimental autoimmune thyroiditis. J Pathol 2008;216:505-513. [ Links ]
53. Ikeda T, Hirata S, Fukushima S, Matsunaga Y, Ito T, Uchino M, Nishimura Y, Senju S. Dual effects of TRAIL in suppression of autoimmunity: the inhibition of TH1 cells and the promotion of regulatory T cells. J Immunol 2010;185:5259-5267. [ Links ]
54. Xiao H, Wang S, Miao R, Kan W. TRAIL is associated with impaired regulation of CD4+CD25- T cells by regulatory T cells in patients with rheumatoid arthritis. J Clin Immunol 2011 July 6 – Epub ahead of print. [ Links ]
55. Yu X, Li L, Li Q, Zang X, Liu Z. TRAIL and DR5 promote thyroid follicular cell apoptosis in iodine excess-induced experimental autoimmune thyroiditis in NOD mice. Biol Trace Elem Res 2011;143:1064-1076. [ Links ]
56. Bretz JD, Mezosi E, Giordano TJ, Gauger PG, Thompson NW, Baker JR Jr. Inflammatory cytokine regulation of TRAIL-mediated apoptosis in thyroid epithelial cells. Cell Death Diff 2002;9:274-286. [ Links ]
57. Spiegel S, Milstein S. Sphingosine-1-phosphate, a key cell signaling molecule. J Biol Chem 2002;277:25851-25854. [ Links ]
58. Olivera A, Rivera J. Sphingolipids and balancing of immune cell function: lessons from the mast cell. J Immunol 2005;174:1153-1158. [ Links ]
59. Hughes JE, Srinivasan S, Lynch KR, Proia RL, Ferdek P, Hedrick CC. Sphingosine-1-phosphate induces an anti-inflammatory phenotype in macrophages. Circ Res 2008;102:950-958. [ Links ]
60. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent of S1P receptor 1. Nature 2004;427:355-360. [ Links ]
61. Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, Chi H. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol 2009;10:769-777. [ Links ]
62. Arendt CW, Albrecht B, Soos TJ, Littman DR. Protein kinase C-theta: signaling from the center of the T-cell synapse. Curr Opin Immunol 2002;14:323-330. [ Links ]
63. Gupta S, Manicassamy S, Vasu C, Kumar A, Shang W, Sun Z. Differential requirement of PKC-θ in the development and function of natural regulatory T cells. Mol Immunol 2008;46:213-224. [ Links ]
64. Zanin-Zhorov A, Ding Y, Kumari S, Attur M, Hippen KL, Brown M, Blazar BR, Abramson SB, Lafaille JJ, Dustin ML. Protein Kinase C-θ mediates negative feedback on regulatory T cell function. Science 2010;328:372-376. [ Links ]
65. Kwon MJ, Wang R, Ma J, Sun Z. PKC-θ is a drug target for prevention of T cell-mediated autoimmunity and allograft rejection. Endocr Metab Immune Disord Drug Targets 2010;10:367-372. [ Links ]
66. Jin Y, Chen X, Podolsky R, Hopkins D, Makala LHC, Muir A, She JX. APC dysfunction is correlated with defective suppression of T cell proliferation in human type 1 diabetes. Clin Immunol 2009;130:272-279. [ Links ]
67. Sharma RB, Fan X, Caturegli P, Rose NR, Burek CL. Invariant NKT cell lines derived from the NODH2h4 mouse enhance autoimmune thyroiditis. J Thyroid Res 2011;2011:895923. [ Links ]
68. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009;31:331-341. [ Links ]
69. Shi Y, Wang H, Su Z, Chen J, Xue Y, Wang S, Xue Y, He Z, Yang H, Zhou C, Kong F, Liu Y, Yang P, Lu L, Shao Q, Huang X, Xu H. Differentiation imbalance of Th1/Th17 in peripheral blood mononuclear cells might contribute to pathogenesis of Hashimoto´s thyroiditis. Scand J Immunol 2010;72:250-255. [ Links ]
70. Nanba T, Watanabe M, Inoue N, Iwatani Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto´s disease and in the proportion of Th17 cells in intractable Graves´ disease. Thyroid 2009;19:495-501. [ Links ]
71. Watanabe M, Yamamoto N, Maruoka H, Tamai H, Masuzuka F, Miyauchi A, Iwatani Y. Independent involvement of CD8+CD25+ cells and thyroid autoantibodies in disease severity of Hashimoto´s disease. Thyroid 2002;12:801-808. [ Links ]
72. Ouyang W, Yang C, Liu Y, Xiong J, Zhang J, Zhong Y, Zhang G, Zhou F, Zhou Y, Xie C. Redistribution of DR4 and DR5 in lipid rafts accounts for the sensitivity to TRAIL in NSCLC cells. Int J Oncol 2011;39:1577-1586. [ Links ]
73. Stassi G, DeMaria R. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat Rev Immunol 2002;2:195-204. [ Links ]
74. Bergelin N, Blom T, Heikkilä J, Löf C, Alam C, Balthasar S, Slotte JP, Hinkkanen A, Törnquist K. Sphingosine kinase as an oncogene: autocrine sphingosine-1-phosphate modulates L-1 thyroid carcinoma cell migration by mechanism dependent on protein kinase C-alpha and ERK1/2. Endocrinology 2009;150:2055-2063. [ Links ]
75. Sekiguchi M, Iwasaki T, Kitano M, Kuno H, Hashimoto N, Kawahito Y, Azuma M, Hla T, Sano H. Role of sphingosine-1-phosphate in the pathogenesis of Sjögren´s syndrome. J Immunol 2008;180:1921-1928. [ Links ]

