








Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
Print version ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. vol.13 no.2 Mérida June 2015
Diagnóstico y manejo del hipogonadismo masculino
Protocolo del Servicio de Endocrinología del Instituto Autónomo Hospital Universitario de Los Andes.
Cesar Escalante, Mariarlenis Lara, Roald Gómez-Pérez, Yajaira Briceño, Grupo de Endocrinología Mérida (ENDO-MER).
Unidad de Endocrinología, Instituto Autónomo Hospital Universitario de los Andes (IAHULA), Mérida, Venezuela.
Dirigir correspondencia a: César Escalante. Email: cesarescalante1@gmail.com
RESUMEN
El hipogonadismo masculino se caracteriza por disfunción testicular y/o del eje hipotálamo-hipofisario produciendo una reducción de las concentraciones de testosterona. Se clasifica en hipogonadismo hipogonadotrópico (Hh) e hipogonadismo hipergonadotrópico (HH), y a su vez en causas adquiridas y congénitas. Las gonadotropinas pueden estar elevadas (HH) o disminuidas (Hh) y en general cursan con niveles disminuidos de testosterona. Dentro de las pruebas que apoyan al diagnóstico y manejo se incluyen el ecosonograma testicular, evaluación genética, densitometría ósea, biopsia testicular, estudios imagenológicos, espermograma, anticuerpos antiespermáticos y las pruebas dinámicas. Por último, la terapia de reemplazo con testosterona es el principal tratamiento, y la meta es obtener valores de testosterona total entre 400 y 700 ng/dL. A continuación, se presenta el protocolo para el diagnóstico y manejo del hipogonadismo masculino, con la evidencia científica y la experiencia clínica de la Unidad de Endocrinología del IAHULA.
Palabras claves: Hipogonadismo masculino, hipogonadismo hipogonadotrópico, hipogonadismo hipergonadotrópico, testosterona.
Male hypogonadism management.
ABSTRACT
Male hypogonadism is characterized by testicular dysfunction secondary to testicular damage and/or hypothalamus-pituitary axis dysfunction resulting in a reduction in testosterone levels. It is classified as hypogonadotropic hypogonadism (Hh) or hypergonadotropic hypogonadism (HH), and these are also divided in acquired and congenital causes. Gonadotropins may be high (HH) or decreased (Hh) and generally occur with decreased testosterone levels. Among the tests supporting the diagnosis and management are included testicular ultrasonography, genetic screening, bone densitometry, testicular biopsy, imaging studies, spermogram, sperm antibodies and hormonal dynamic tests. Finally, testosterone replacement therapy is the main treatment, and the goal is to obtain total testosterone values between 400 and 700 ng/dL. Below, is presented the IAHULA Endocrinology Unit guidelines for male hypogonadism management, based on scientific evidence and clinical experience.
Keywords: Male hypogonadism, hypogonadotropic hypogonadism, hypogonadotropic hypogonadism, testosterone.
Articulo recibido en: Febrero 2015 Aceptado para publicación en: Mayo 2015
GENERALIDADES
El hipogonadismo masculino está caracterizado por una disfunción testicular la cual puede afectar la espermatogénesis y/o síntesis de testosterona. Puede resultar de un daño testicular o por una disfunción en el eje hipotálamo-hipofisario1, produciendo una reducción de las concentraciones de testosterona2. Según sea el origen del defecto, se puede clasificar en hipogonadismo hipogonadotrópico (Hh) cuando es a nivel hipotálamo/hipofisario o bien en hipogonadismo hipergonadotrópico (HH) cuando el defecto es de la gónada per se. La prevalencia en general de esta enfermedad aún es desconocida1. El síndrome de Klinefelter es la manifestación congénita de HH más común y afecta 1 de cada 500 hombres, mientras que en el caso del Hh, la prevalencia está de 1:10000 a 1:86000 casos1. Si se considera la deficiencia androgénica asociada a envejecimiento, la prevalencia de hipogonadismo se eleva significativamente3.
FISIOLOGÍA
El hipotálamo, la hipófisis y los testículos forman un sistema integrado que es responsable de la secreción adecuada de las hormonas masculinas y la espermatogénesis. Los testículos requieren la estimulación por parte de la hormona folículo estimulante (FSH) y hormona luteinizante (LH) que son secretadas en respuesta a la hormona liberadora de gonadotropina hipotalámica (GnRH). El pulso generador de GnRH es el principal regulador de la pubertad y su producción comienza temprano en la vida fetal, cambiando los niveles de gonadotropina drásticamente en periodo fetal, infancia, pubertad y adultez. Mientras que la FSH actúa directamente sobre el epitelio germinal, la LH estimula la secreción de testosterona en las células de Leydig. Esta última, estimula la virilización, la producción espermática y realiza la retroalimentación hipotálamo/hipofisario para regular la secreción de GnRH1.
En cuanto a la testosterona, solo 1 a 2% circula libre en sangre y el restante 98 a 99% está unido a la albumina en un 40-50% y a la globulina trasportadora de hormonas sexuales (SHBG) en un 50-60%. La testosterona libre y la unida a la albumina conforman la llamada testosterona biodisponible, la cual es medible a nivel sérico. Finalmente, la testosterona puede ejercer acción directamente en su receptor de andrógeno, o bien metabolizarse para formar dihidrotestosterona (DHT) (metabolito activo), convertirse en metabolitos con menor actividad biológica y/o producir estradiol (E2) (Tabla I, Fig. 1)4-7.

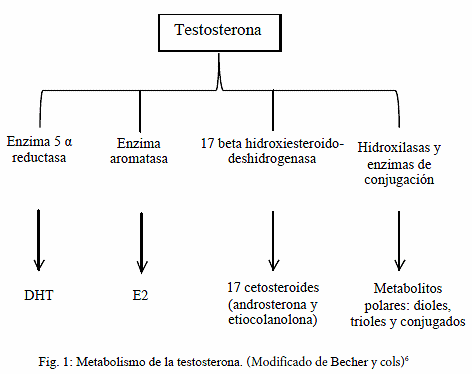
En la fisiopatología del hipogonadismo, el déficit hormonal en los casos de Hh puede ser hipotalámico o hipofisario, parcial o total e incluir alteración en los niveles de LH y FSH o en solo una de ellas, y en algunos casos, estar asociado con alteraciones de otras hormonas hipofisarias. En las formas congénitas, en los casos leves puede haber falta del desarrollo puberal, y en los casos más graves, defectos en los genitales como criptorquidia, micropene o ambigüedad genital. En los HH, a partir de los 9 a 10 años de edad, la acción de los esteroides sexuales está reducida, y al no lograr la retroalimentación negativa hipofisaria, aumenta la producción de gonadotropinas pero sin lograr la normalización de la función gonadal8.
CLASIFICACIÓN
Como ha sido descrito anteriormente, todas las causas de hipogonadismo masculino se resumen en dos grandes grupos, el HH y el Hh. No obstante, otros tipos de hipogonadismo han sido reportados como resultado de una disminución de la sensibilidad o insensibilidad a la testosterona y sus metabolitos, o por reducción de la biodisponibilidad secundario a un incremento de la SHBG9. A continuación se describen detalladamente las distintas etiologías del hipogonadismo.
1.-Hipogonadismo hipergonadotrópico (HH-Primario).
Es la forma más frecuente de hipogonadismo en el hombre adulto. Esta caracterizado por disminución en la producción de testosterona y elevación de LH y FSH1. Puede deberse a causas congénitas como anormalidades cromosómicas, síndromes o mutaciones genéticas. También puede ser adquirido en la niñez o adolescencia por autoinmunidad o exposición a quimioterapia o radiación10,11.
1.1.- Causas Congénitas.
El síndrome de Klinefelter es la causa más frecuente. El genotipo más común es 47XXY (75%) seguido de 46XY/47XXY (20%) y otras variantes (5%)12. Las características clínicas distintivas son talla alta, hábito eunucoide, testículos firmes y pequeños y ginecomastia. La disgenesia de los túbulos seminíferos es una característica clásica y el espectro de la falla gonadal puede variar desde pubertad retrasada en la adolescencia, falta de adecuada progresión puberal o infertilidad en la adultez10.
Otras causas de HH congénito son mutaciones de la subunidad beta de las gonadotropinas o de sus receptores. En efecto, los varones con mutaciones de la subunidad beta de la FSH pueden tener pubertad normal o retrasada con azoospermia y los que tienen mutación de la subunidad beta de LH pueden cursar con pérdida total de la función gonadal. Otra etiología es el testículo evanescente, donde existen genitales externos masculinos normales que denotan una función testicular normal en vida fetal, pero luego los testículos se atrofian, por torsión testicular antenatal o trauma en el contenido escrotal in útero10.
La criptorquidia es la alteración genital más frecuentemente observada en el recién nacido, y se incluye como otra causa de hipogonadismo. La incidencia es de 2,2 a 3,8% en recién nacidos a término y de 0,7 a 1,3% después del primer año de vida. El daño testicular es frecuente en la criptorquidia y se plantean 3 mecanismos fisiopatológicos. El primero es una anomalía testicular primaria (gónada disgenética), el segundo es un estado de Hh transitorio por falla en el incremento de las gonadotropinas posterior al nacimiento, causando involución de células de Leydig, y el tercero es la elevación de la temperatura testicular que altera el desarrollo del testículo. En la adolescencia y en el adulto la mayoría de los testículos criptorquídicos presentan anormalidad estructural13.
1.2.- Causas adquiridas.
El envejecimiento se encuentra como causa de hipogonadismo en un 4,1% y un 9,3% en las edades entre 40 a 49 años y 60 a 70 años, respectivamente. Otras de las causas asociadas a hipogonadismo son la castración quirúrgica, traumas, iatrogénicas, torsión bilateral, orquitis (virus de la parotiditis), exposición a agentes gonadotóxicos (quimioterapia y radioterapia) y autoinmunidad1,11,14. En cuanto a los agentes gonadotóxicos, el efecto de la radiación en la función testicular es dependiente de la edad; cuando es pre-puberal se presenta más daño en células de Leydig que en los casos de pacientes en estado post-puberal. En la etapa puberal se ha encontrado que las células de Leydig son más resistentes a los daños de la radioterapia que las células germinales, y hay una progresión normal de la pubertad a pesar del deterioro severo de la espermatogénesis15. Las dosis acumulativas de agentes alquilantes también se correlacionan con una función alterada10.
Por otro lado, en el síndrome poliglandular autoinmune se puede presentar falla gonadal. Si esta ocurre, la tasa de presentación es más baja en hombres que en mujeres. Se ha encontrado que la autoinmunidad en las células de Leydig es mediada por los anticuerpos P450 (específico de testículo) y en los casos de varones pre-puberales tratados con quimioterapia y en aquellos con anormalidades de tracto urogenital como criptorquidia, torsión testicular e hipospadia, el anticuerpo observado es el anti-espermático10.
La orquitis autoinmune es definida como una agresión autoinmune donde existen anticuerpos específicos antiespermáticos (ASA) positivos, produciendo anormalidades espermáticas e infertilidad. En estos casos no existe anormalidad hipotálamo-hipofisaria y las hormonas gonadales pueden estar en rango normal. Existen dos tipos de orquitis, la primaria caracterizada por infertilidad y presencia de ASA en un 100% de los casos, sin la evidencia de una enfermedad sistémica, y la secundaria, caracterizada por vasculitis testicular en asociación con una enfermedad sistémica autoinmune (presencia de ASA en un 50% y 20% en lupus eritematosos sistémico y artritis reumatoide, respectivamente)16.
2.-Hipogonadismo hipogonadotrópico (Hh- Secundario)
Es una consecuencia de enfermedades que afectan la secreción de GnRH o LH/FSH, encontrándose disminuidas o ausentes1. Puede ser atribuido de una variedad de defectos congénitos como mutaciones puntuales, síndromes genéticos y formas idiopáticas. La causa más frecuente es transitoria y se llama retardo constitucional del crecimiento y desarrollo10. Cada una de estas causas es brevemente discutida en esta revisión (Tabla II).

2.1.- Causas Congénitas
Se clasifican dependiendo de la presencia o no de alteración del sentido del olfato. La incidencia es de 1-10:100.000 nacidos vivos, con una relación para Hh anósmico (síndrome de Kallman) de 2/3 y para Hh aislado con sentido del olfato normal (Hh idiopático) de 1/3 de los casos. En cuanto a esta última patología, los niveles bajos de gonadotropinas y hormonas sexuales se han observado en ausencia de anormalidades anatómicas del eje hipotálamo/hipófisis/gonadal y se plantea que la falla en la diferenciación o desarrollo de las neuronas de GnRH, son las causantes de la falta de secreción o secreción inadecuada (no pulsátil) de esta hormona1. En otros casos, se presentan defectos en el receptor de GnRH10.
En cuanto al Hh anósmico (síndrome de Kallman), se produce por mutaciones del gen KAL-1 que conlleva a un desorden en la migración de las neuronas de GnRH y olfatorias. La falla de las neuronas de GnRH para migrar desde la placa olfatoria a su destino en el hipotálamo y en el lóbulo olfatorio, representa el defecto embriológico básico de este síndrome1. Clínicamente puede presentar sinquinesia, agenesia de cuerpo calloso, trastorno visuoespacial, ptosis palpebral congénita, alteraciones auditivas, labio o paladar hendido, hipodontia, agenesia renal unilateral, alteraciones estructurales en dedos de manos o pies, obesidad y azoospermia17. Se han encontrado casos que dentro de una misma familia existen individuos con Hh aislado y otros con síndrome de Kallman10. Otro síndrome a mencionar es el síndrome de Laurence-Moon-Biedl, menos frecuente, autosómico recesivo y puede cursar con 5 síntomas principales: hipogonadismo, retinitis pigmentosa, polidactilia/sindactilia, obesidad y oligofrenia6,18.
Mutaciones de receptores nucleares como el factor esteroidogénico -1 (SF-1), un regulador involucrado en la diferenciación sexual, esteroidogénesis y reproducción, puede conllevar a disgenesias testiculares y falla adrenal en genotipo masculino. Otro caso, es la mutación DAX-1 que típicamente presentan disfunción hipotalámico/hipofisaria, defectos en la espermatogénesis, insuficiencia adrenal de inicio temprano y subsecuente pubertad retrasada. Se ha planteado que la mutación de estos receptores nucleares puede llevar a hipogonadismo de múltiples formas10,19. En el Hh congénito se pueden encontrar defectos en el gen Prop-1 (produce deficiencia hormonales hipofisarias), defectos del receptor de leptina (produce supresión del eje reproductivo) y otros síndromes como Prader-Willi (produce disfunción hipotalámica)10.
2.2.- Causas Adquiridas.
Cualquier lesión significativa del sistema nervioso central puede resultar en Hh adquirido10. Algunas de las condiciones que producen esta patología son: drogas, fármacos (esteroides sexuales, análogos de GnRH, corticosteroides, opioides) enfermedades infiltrativas e infecciosas hipofisarias, tumores hipofisarios (13% pueden tener anormalidades en la secreción de gonadotropinas antes de inicio de la terapia), hiperprolactinemia, panhipopituitarismo, traumatismo encefálico (se puede encontrar en el 90 a 95% de los pacientes con historia de trauma cerebral), radiación hipofisaria cerebral, ejercicio extremo, abuso de alcohol y enfermedades sistémicas (hemocromatosis, sarcoidosis, histiocitosis, enteropatías, cirrosis hepática, enfermedad renal, diabetes, drepanocitosis, enfermedad tiroidea, hipercortisolismo, síndrome de inmunodeficiencia adquirida, tuberculosis, distrofia miotónica de Steiner, epilepsia)1,10,18.
DIAGNÓSTICO
Es importante realizar una anamnesis completa que incluya: inicio de síntomas, antecedentes personales (fracturas, cirugía testicular o sellar, fármacos), antecedentes familiares (padre con pubertad retrasada, antecedentes de hipogonadismo familiar) e historia sexual (frecuencia de coito, deseo sexual, erecciones matutinas espontáneas)20.
Al examen físico evaluar: índice de masa corporal, presión arterial, proporciones corporales (hábito eunucoide), ginecomastia, virilización, estadio de pubertad según Tanner, posición de meato uretral (hipospadia, epispadia), pliegues y pigmentación del escroto, tamaño y consistencia testicular (tumores, hidrocele, varicocele, espermatocele) y evaluación prostática20. En la Tabla III se resumen algunas de las manifestaciones sugestivas de hipogonadismo.
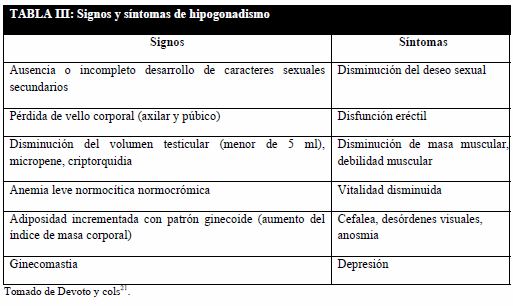
Otras condiciones en las cuales se debe sospechar hipogonadismo son, masa ósea baja (fractura de bajo impacto, osteoporosis), diminución de la frecuencia del rasurado, enfermedades de la región sellar, uso de fármacos (opioide, glucocorticoides), pérdida de peso asociada a VIH, enfermedad renal crónica estadio V, enfermedad pulmonar obstructiva crónica moderada a severa, infertilidad y síndromes metabólicos (diabetes y obesidad)22,23.
LABORATORIO
El diagnóstico de hipogonadismo se basa en niveles de testosterona total menor de 300 ng/dL por lo menos en dos oportunidades, con al menos 1 signo o síntoma clínico previamente mencionado14. Si los valores de testosterona total son normales o se encuentran en el límite inferior de lo normal, se deberá realizar SHBG y testosterona libre. Es importante a su vez solicitar concentraciones séricas de LH, FSH y prolactina para evaluar la indemnidad hipofisaria (Tabla IV). Bajos niveles de gonadotropinas más niveles bajos de testosterona son indicativos de Hh y en el caso de encontrar altos niveles de gonadotropinas y niveles bajos de testosterona son indicativos de HH24. Se deben incluir otros exámenes de laboratorio para descartar enfermedades sistémicas (hematología completa, función tiroidea, renal y hepática).

PRUEBAS COMPLEMENTARIAS
Dentro de las pruebas que apoyan al diagnóstico y manejo de hipogonadismo se incluyen: ecosonograma testicular en paciente con hallazgos de masas testiculares o escrotales y para evaluar tamaño testicular; evaluación genética (cariotipo) en pacientes con HH mas desarrollo puberal inapropiado y ginecomastia, donde se sospeche un síndrome de Klinefelter; densitometría ósea para evaluación del riesgo o presencia de masa ósea baja secundario a déficit de hormonas sexuales; biopsia testicular solo en azoospermia secundaria a anormalidades de los vasos o de las células germinales y/o obstrucciones que presente FSH normal y volumen testicular normal; estudios imagenológicos (RMN silla turca) para evaluar estructura hipofisaria en caso de Hh no atribuible claramente a una causa específica, cuando existen niveles de testosterona ≤ 150 ng/dL y/o hiperprolactinemia no explicada; espermograma para estudio de fertilidad (diagnóstico de azoospermia); anticuerpos antiespermáticos como parte del estudio inmunológico24.
PRUEBAS DINÁMICAS
Prueba de Gonadotropina Coriónica Humana (HCG): Es una prueba de función testicular que se utiliza para diferenciar entre criptorquidia y anorquia en caso de que se sospeche de HH. Se determina testosterona basal, se administra HCG en forma intramuscular (500 UI en menores de 2 años, 1000 UI en niños de dos a cuatro años, 1500 UI hasta los 12 años, 2500 UI en edad puberal y 5000 UI en adultos), y luego de 48 a 72 horas se toma una segunda muestra. La interpretación en los lactantes es que la testosterona total debe aumentar 2 a 10 e incluso 20 veces el valor basal, en la niñez debe aumentar 5 a 10 veces, en la pubertad 2 a 3 veces y en los adultos aumentar 1,5 a 2 veces el valor basal, confirmando la presencia de tejido testicular20,25-27.
Prueba de estímulo con GnRH: Se administra un bolo endovenoso de 100 μg de GnRH en el adulto y se mide LH y FSH basal, a los 30, 45 y 60 minutos. La respuesta normal es la elevación de LH 3 a 6 veces el valor basal y un 20 a 50% el valor de FSH. Según el grado de insuficiencia testicular primaria se encontrarán niveles más elevados de los esperados de LH y FSH. En los casos de individuos con enfermedad hipofisaria y/o hipotalámica, pueden tener una respuesta normal o reducida de dichos valores, no obstante, esta prueba en insuficiente para distinguir entre una y otra24.
Prueba de estimulación con clomifeno: La base para el uso de este fármaco es que actúa interrumpiendo la retroalimentación negativa de las hormonas sexuales y estimula la liberación de gonadotropinas de la hipófisis. Se administra 100 mg de acetato de clomifeno en forma oral por 5 a 7 días. Se mide LH y FSH basal y entre el 5° y 7° día; si incrementa el nivel de LH 2 veces el valor basal y el nivel de FSH en un 20 a 50%, es indicativo de función hipotalámica e hipofisaria normal24.
TRATAMIENTO
La terapia de reemplazo con testosterona es la opción principal del tratamiento para hipogonadismo masculino28. Idealmente la terapia debería proveer niveles fisiológicos de testosterona14 y luego de la pubertad, no hay límite de edad para iniciar su aplicación. La testosterona corrige el cuadro clínico entre 1 a 2 meses de iniciada, y la restitución total puede necesitar más tiempo3. A continuación, se muestran los fármacos empleados para la restitución hormonal (Tabla V)22,29,31.

Metas en la terapia de reemplazo con testosterona.
Las metas establecidas en el tratamiento de la testosterona son22,24.
Testosterona total en valores entre 400 y 700 ng/dL.
Mejoría de signos y síntomas.
Producir y mantener virilización.
Optimizar densidad ósea y prevenir osteoporosis.
Restaurar fertilidad en caso de Hh.
Precauciones y efectos adversos en la terapia de reemplazo con testosterona.
Previo al inicio del tratamiento con testosterona se debe evaluar hematocrito basal, a los 3 meses y luego anual. Si el hematocrito es > 54%, se debe detener la terapia hasta que éste disminuya a un nivel de seguridad. Luego reiniciar la terapia con una menor dosis. En caso de presentar cualquiera de las siguientes situaciones: elevación de antígeno prostático específico (PSA) > 1,4 ng/mL en cualquier momento, en un periodo de 12 meses, una velocidad de aumento del PSA > 0,4 ng/mL/año (utilizar el PSA medido al 6° mes de iniciada la testosterona como referencia), detección de anormalidad prostática al tacto rectal o un índice AUA/IPSS (American Urological Association symptom score/International Prostate Symptom Score) >19, se debe solicitar evaluación por urología. Es importante a su vez, no solo evaluar hematocrito y próstata, sino también evaluar otros posibles efectos adversos de la testosterona, los cuales se resumen en la Tabla VI22.

Las recomendaciones para la monitorización de la terapia de restitución con testosterona son31:
Inicio:
Evaluar próstata con examen físico y determinación de PSA sobre todo en hombres > 45 años de edad.
Determinar testosterona sérica.
Determinar el hematocrito, las transaminasas y la bilirrubina.
Evaluar densidad mineral ósea.
Cada 3 meses en el primer año de inicio de tratamiento:
Niveles de testosterona a los 3 meses del inicio del tratamiento:
Enantato de testosterona intramuscular: medir a mitad del intervalo entre una inyección y otra.
Undecanoato de testosterona intramuscular: medir justo antes de cada inyección.
Parches: medir a las 3 a 12 horas posterior a su aplicación.
Tabletas bucales: medir inmediatamente previa aplicación o posterior a la misma.
Gel: después de 1 semana de uso, medir en cualquier momento.
Revisión prostática y determinación de PSA.
Determinar hematocrito.
Determinar efectos adversos de la testosterona.
Anual:
Revisión prostática y del hematocrito.
Evaluar efectos adversos de testosterona.
Cada 2 años:
Revisión de la densidad mineral ósea.
La testosterona esta contraindicada en cáncer de próstata o mamas, nódulos o induración prostática palpable, antígeno prostático mayor de 4 ng/mL, hematocrito mayor de 50%, apnea obstructiva del sueño severa y no controlada, síntomas severos de obstrucción del tracto urinario bajo, insuficiencia cardiaca mal controlada.
En la Tabla VII se resumen éstas contraindicaciones según el riesgo22.


Terapia de reemplazo con testosterona y riesgo cardiovascular.
Si bien múltiples estudios sugieren que los niveles de testosterona normales juegan un papel importante en el mantenimiento de la salud cardiovascular, como se ha demostrado en algunos casos con la terapia de reemplazo de testosterona en hipogonadismo, que reportan mejoría de la isquemia miocárdica, la capacidad de ejercicio, la longitud de segmento QTc, la diabetes y la obesidad32, otros estudios comentan que el hallazgo de aumento del riesgo cardiovascular en pacientes con dicha terapia ha sido destacado en los últimos años. En el año 2010 un pequeño ensayo aleatorio con gel de testosterona en hombres mayores de 65 años de edad, fue suspendido, debido a un exceso de una variedad de eventos cardiovasculares33,34. Otro estudio en el 2014 encontró que una vez iniciada la terapia con testosterona en los primeros 90 días, el riesgo de eventos coronarios era 2 a 3 veces mayor, demostrando que los hombres jóvenes que tienen antecedentes de enfermedad coronaria tenían un mayor riesgo de presentar eventos cardiovasculares. El riesgo relativo (RR) para hombres menores de 65 años fue de 1.17 (0.84, 1.63) y en hombres mayores de 65 años de edad fue de 2.19 (1.27, 3.77)33. En la actualidad, una revisión sistemática no encontró estudios aleatorizados, ni controlados con placebo sobre los efectos de la terapia con testosterona sobre enfermedad cardiovascular en jóvenes hipogonádicos21 y se necesitan más estudios con poder estadístico para aclarar si la intervención hormonal reduciría la incidencia de las enfermedades cardiovasculares en hombres de mediana y avanzada edad35. La Sociedad Americana de Endocrinología en su guía de manejo, no recomienda el tratamiento de pacientes con enfermedades cardiacas para mejorar la supervivencia33. Por lo tanto, se sugiere que previo inicio del tratamiento de terapia de reemplazo con testosterona independientemente de la edad, debería realizarse una evaluación cardiovascular.
REFERENCIAS BIBLIOGRÁFICAS
1. Fraietta R, Suslik D, Esteves S. Hypogonadotropic hypogonadism Revisited. Clinics (Sao Paulo) 2013; 68 (Suppl 1):81-88. [ Links ]
2. Mulligan T, Frick M, Zuraw Q, Stemhagen A, Mcwhirter C. Prevalence of hypogonadism in males aged at least 45 years: the HIM study. Int J ClinPract 2006;60: 762769. [ Links ]
3. Knoblovits P, Levalle O, Nagelberg A, Pacenza N, Rodríguez M. Segundo Consenso Argentino sobre Patologías Endocrinológicas Área Hipogonadismo masculino. RAEM 2007;44:133-145. [ Links ]
4. Dandona P, Rosenberg M. A practical guide to male hypogonadism in the primary care setting. Int J ClinPract 2010;64:682-696. [ Links ]
5. Jockenhövel F, Schubert M. Anatomy and physiology of the testi. En: Jockenhövel F, Schubert M. Male Hypogonadism. 3rd edition. Bremen: UNI-MED Verlag AG Science 2009: pp -11-31.
6. Becher E, Torres L, Glina S. Consenso Latinoamericano sobre DAEM. Primera edición. Sao Paulo: Planmark 2013.
7. Pantalone K, Faiman C. Male hypogonadism: More than just a low testosterone. Cleveland Clin J Med 2012;79:717-725. [ Links ]
8. Rodriguez F. Pubertad retrasada e hipogonadismos. En: Argente A, Carrascosa A, Gracia R, Rodríguez F. Tratado de Endocrinología Pediátrica y de la Adolescencia. Segunda Edición. Ediciones Doyma. Barcelona 2000: 883-911.
9. Corona G, Rastrelli G, Vignozzi L, Mannucci E, Maggi M. How to recognize late-onset hypogonadism in men with sexual dysfunction. Asian J Androl 2012;14: 251259. [ Links ]
10. Viswanathan V, Eugster E. Etiology and treatment of hypogonadism in adolescents. Pediatr Clin North Am 2011;58:11811200. [ Links ]
11. Martul P. Hipogonadismo: diagnóstico y tratamiento. An Esp Pediatr 2000;52:59-62. [ Links ]
12. Jubiz W, Cruz E. Hipogonadismo masculino: Causas, genética, diagnóstico y tratamiento. Colomb Med 2007; 38:84-89. [ Links ]
13. Gómez R. Criptorquidia: Importancia del diagnóstico y tratamiento precoz. Rev Venez Endocrinol Metab 2004; 2:14-17. [ Links ]
14. Kumar P, Kumar N, Thakur D, Patidar A. Male hypogonadism: symptoms and treatment. J Adv Pharm Technol Res 2010;1:297-301. [ Links ]
15. Wallace W, Thomson A. Preservation of fertility in children treated for cancer. Arch Dis Child 2003;88: 493496. [ Links ]
16. Silva A, Cocuzza M, Carvalhod J, Bonfá E. Diagnosis and classification of autoimmune orchitis. Autoimmun Rev 2014;13:431434. [ Links ]
17. Gutiérrez B, Figuera L, Orozco R. Síndrome de Kallmann. Aspectos genéticos y variantes fenotípicas. Rev Med Inst Mex Seguro Soc 2012; 50:157-161. [ Links ]
18. Jockenhövel F, Schubert M. Hypogonadism in men. En: Jockenhövel F, Schubert M. Male Hypogonadism. 3rd edition. Bremen: UNI-MED Verlag AG Science 2009: pp -33-81.
19. Jadhav U, Harris R, Jameson L. Hypogonadotropic hypogonadism in subjects with DAX1 mutations. Mol Cell Endocrinol 2011;346:6573.
20. Jockenhövel F, Schubert M. Diagnostic work-up of hypogonadism. En: Jockenhövel F, Schubert M. Male Hypogonadism. 3rd edition. Bremen: UNI-MED Verlag AG Science 2009: pp -83-91.
21. Devoto E, Aravena L. Hipogonadismo asociado a la senilidad en el varón (climaterio masculinoandropausia adam). Rev Chil Obstet Ginecol 2004; 69:392-398.
22. Bhasin S, Cunningham G, Hayes F, Matsumoto A, Snyder P, Swerdloff R, Montori V. Testosterone therapy in men with androgen deficiency syndromes: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2010. 95:2536-2559.
23. National Kidney Foundation. Clinical Practice Guidelines for chronic kidney disease: evaluation, classification and stratification. Am J Kidney Dis 2002;39: s1-266.
24. Anderson R, Bergman D, Garber J, Hamilton C, Hershon K, Jovanovic L, Kleerekope M, Mechanick J, Palumbo P, Peters A, Rettinger H, Rodbard H, Rubenstein H, Seibel J, Tourtelot J. American Association of Clinical Endocrinologists Medical Guidelines for clinical practice for the evaluation and treatment of hypogonadism in adult male patients--2002 update. Endocr Pract 2002; 8:439-456.
25. Lechuga J, Lechuga A. Criptoquidia. Protoc Diagn Ter Pediatr 2011;1:1:34-43.
26. Argente J, Carrascosa A, Gracia R, Rodriguez F. Pruebas funcionales en endocrinologia pediátrica y de la adolescencia. En: Argente A, Carrascosa A, Gracia R, Rodríguez F. Tratado de Endocrinología Pediátrica y de la Adolescencia. Segunda Edición. Ediciones Doyma. Barcelona 2000:1425.
27. Degros V, Cortet-Rudelli C, Soudan B, Dewailly D. The human chorionic gonadotropin test is more powerful than the gonadotropin-releasing hormone agonist test to discriminate male isolated hypogonadotropic hypogonadism from constitutional delayed puberty. Eur J Endocrinol 2003;149:2329.
28. Surampudi P, Wang C, Swerdloff R. Hypogonadism in the aging male diagnosis, potential benefits, and risks of testosterone replacement therapy. Int J Endocrinol 2012;2012:625434.
29. Jockenhövel F, Schubert M. Androgen treatment options. En: Jockenhövel F, Schubert M. Male Hypogonadism. 3rd edition. Bremen: UNI-MED Verlag AG Science 2009: pp -93-119.
30. Yassin A, Haffejee M. Testosterone depot injection in male hypogonadism: a critical appraisal. Clin Interv Aging 2007;2:577590.
31. Becerra A, Acosta L. Documento básico de consenso sobre el síndrome de Hipogonadismo de inicio tardío. Endocrinol Nutr 2008;55:5-28.
32. Oskui P, French W, Herring M, Mayeda G, Burstein S, Kloner R. Testosterone and the cardiovascular system: A comprehensive review of the clinical literature. J Am Heart Assoc 2013;2:e000272.
33. Finkle W, Greenland S, Ridgeway G, Adams J, Frasco M, Cook M, Fraumeni JF Jr, Hoover RN. Increased risk of non-fatal myocardial infarction following testosterone therapy prescription in men. PLoS One 2014;9: e85805.
34. Basaria S, Coviello AD, Travison TG, Storer TW, Farwell WR, Jette A, Eder R, Tennstedt S, Ulloor J, Zhang A, Choong K, Lakshman K, Mazer N, Miciek R, Krasnoff J, Elmi A, Knapp P, Brooks B, Appleman E, Aggarwal S, Bhasin G, Hede-Brierley L, Bhatia A, Collins L, LeBrasseur N, Fiore L, Bhasin S. Adverse events associated with testosterone administration. N Engl J Med 2010;363:109122.
35. Yeap B. Sex steroids and cardiovascular disease. Asian J Androl 2014;16:239247.
36. Chan I, Tang M, Zajac J, Grossmann M. Assessment and management of male androgen disorders: an update. AFP 2014;43:277-282.