










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. vol.15 no.1 Mérida feb. 2017
Revisión
POSIBLES RELACIONES ENTRE ENFERMEDAD DE ALZHEIMER, INSULINORRESISTENCIA Y DIABETES MELLITUS TIPO 2
Belinda Hómez.
Unidad de Endocrinología, Centro Médico Paraíso, Maracaibo, Estado Zulia.
RESUMEN POSIBLES RELACIONES ENTRE ENFERMEDAD DE ALZHEIMER, INSULINORRESISTENCIA Y DIABETES MELLITUS TIPO 2
Belinda Hómez.
Unidad de Endocrinología, Centro Médico Paraíso, Maracaibo, Estado Zulia.
La Enfermedad de Alzheimer (EA) es la causa más común de demencia. Existen múltiples factores de riesgo asociados con la enfermedad, entre ellos la diabetes mellitus tipo 2. En los últimos años un cuerpo creciente de evidencia involucra la insulinorresistencia como causa y consecuencia del desarrollo de lesiones cerebrales típicas de la EA. Actualmente el diagnóstico de EA se basa en criterios clínicos neuropsiquiátricos y confirmación histopatológica post morten; sin embargo, grandes avances en pruebas genéticas, marcadores bioquímicos y neuro-imágenes funcionales permitirán aumentar la precisión diagnóstica. La asociación de alteraciones en la vía de señalización de la insulina con la EA ha dado origen al interés de evaluar agentes originalmente desarrollados para el tratamiento de la diabetes tipo 2 como potenciales terapias. Sin duda aún se requieren numerosas investigaciones que permitan consolidar los hallazgos hasta ahora encontrados.
Palabras clave: Enfermedad de Alzheimer, insulinorresistencia, diabetes mellitus tipo 2.
POSSIBLE RELATIONSHIPS BETWEEN ALZHEIMER’S DISEASE, INSULIN RESISTANCE AND TYPE 2 DIABETES MELLITUS ABSTRACT
Alzheimer’s Disease (AD) is the most common cause of dementia. There are multiple risk factors associated with the disease, including type 2 diabetes mellitus. In recent years a growing body of evidence involves insulin resistance as cause and consequence of the development of brain lesions typical of AD. Currently the diagnosis of AD is based on clinical neuropsychiatric criteria and postmortem histopathological confirmation; however, major advances in genetic testing, biochemical markers and functional neuroimaging will increase diagnostic accuracy. The association of alterations in the signaling pathway of insulin with the AE has given rise to the interest of evaluating agents originally developed for the treatment of type 2 diabetes as potential therapies. Numerous investigations are undoubtedly still needed to consolidate the findings found so far.
Key word: Alzheimer’s disease, insulin resistance, type 2 diabetes mellitus.
INTRODUCCIÓN
La enfermedad de Alzheimer representa el 60-70% de todas las causas de demencia. Según la Organización Mundial de la Salud (OMS)1, la demencia es un síndrome, generalmente crónico o progresivo, caracterizado por el deterioro de la función cognitiva más allá de lo que podría considerarse una consecuencia del envejecimiento normal. Actualmente en el mundo 47,5 millones de personas padecen demencia y se estima que para el año 2050 dicha cifra se triplicará1.
Desde el punto de vista histopatológico lo que caracteriza a la EA es la presencia de placas neuríticas, estas son estructuras redondeadas ubicadas entre las neuronas, constituidas principalmente por el amiloide beta (Aβ) y los ovillos neuro-fibrilares que son lesiones intracelulares cuyo principal componente es la proteína TAU hiper-fosforilada. Ambas lesiones se distribuyen en el parénquima cerebral en densidad y patrón que son característicos de la enfermedad2.
Existen numerosos factores de riesgo que predis- ponen al desarrollo de EA, unos mejor validados que otros, algunos modificables y otros no, entre los que se pueden mencionar: historia familiar de EA, presencia de Apo E4 y otros genes específicos, envejecimiento, bajo nivel educativo, trauma craneal, hábito tabáquico, alcoholismo, sobrepeso, inactividad física, baja estimulación mental y social, diabetes mellitus, hipertensión arterial, hiperlipidemia, enfermedad cardiovascular, depre- sión y ansiedad3. No hay duda de que los más importantes son el envejecimiento y ser portador del alelo Apo E4. Según el metanálisis de Farrer y col4, individuos con este alelo homocigoto presentan un 91% de probabilidad de desarrollar EA, mientras que en los heterocigotos se expresa en un 47%; sin embargo, los individuos no portadores también la pueden desarrollar, de manera que la presencia del alelo Apo E4 no es indispensable ni suficiente para la expresión de la EA, lo que ha llevado a la búsqueda de otros agentes involucrados en su patogenia, entre ellos la insulinorresistencia (IR).
INSULINA Y CEREBRO
El cerebro es el principal consumidor de glucosa, requiriendo dos tercios del total de la glucosa circulante5; aunque históricamente se consideraba al cerebro como un órgano insulino independiente, en los últimos 20 años, creciente evidencia científica apoya el concepto de que la insulina lleva a cabo acciones importantes en el funcionamiento cerebral. La insulina producida en el páncreas atraviesa la barrera hemato-encefálica a través de un sistema de transporte saturable mediado por un receptor de insulina, aunque también existen reportes de producción de insulina en las células cerebrales6. Los receptores de insulina se encuentran ampliamente expresados en el cerebro pero con mayor densidad en la corteza cerebral, hipocampo, hipotálamo y bulbo olfatorio7, y son diferentes en estructura y función a los receptores de insulina en la periferia8. Así tenemos, que la insulina en el cerebro disminuye el apetito, mientras que en la periferia disminuye la glucosa y aumenta el apetito9. También se ha demostrado expresión de glucotransportadores insulinodependientes en células cerebrales10. En el humano, la insulina cumple funciones cerebrales de aprendizaje y memoria, en especial memoria verbal11; estas funciones han sido soportadas por la evidencia que demuestra que la insulina modula la secreción de neurotransmisores como la acetilcolina12 y favorece la plasticidad sináptica13.
INSULINORRESISTENCIA Y ENFER- MEDAD DE ALZHEIMER
Normalmente la insulina inicia sus efectos celulares al unirse al receptor de insulina (RI) ubicado en la superficie celular, lo que desencadena la actividad de la tirosin kinasa que fosforila los miembros de una familia de 4 proteínas, denominadas sustrato del receptor de insulina (SRI 1-SRI 4). Una vez fosforilado en el residuo tirosina (pTir) se acoplan RI-SRI desencadenando una cascada de reacciones que permiten la reprogramación metabólica y transcripcional de la célula14.
En los estados de estrés metabólico y proinflamatorio como la DM, el factor de necrosis tumoral alfa (FNTα), citoquina proinflamatoria, permite la activación aberrante de la c-Jun-N terminal kinasa (JNK) quien fosforila el SRI 1 en el residuo serina (pSer), impidiendo el acoplamiento RI-SRI y de esta manera bloqueando la cascada de señalización de insulina, causando IR periférica14. A la EA se le ha denominado diabetes tipo 3 o forma cerebral de la diabetes, debido a la similitud que se ha encontrado entre los mecanismos de IR periférica que conllevan a DM tipo 2 (DM 2) y la alteración de la señalización de la insulina cerebral observada en la EA15.
El estudio de Rotterdam16 fue el primero en sugerir que sujetos con DM 2 tenían mayor riesgo de desarrollar EA; esta misma conclusión se obtuvo tanto en el estudio asiático de envejecimiento17, como en el metanálisis de estudios epidemiológicos que evaluaron el riesgo de desarrollar EA en sujetos diabéticos18. Numerosos son los potenciales mecanismos etiopatogénicos involucrados en el desarrollo de demencia en la DM 2 además de la IR, como son hiperglucemia crónica, estrés oxidativo, acúmulo de productos finales de la glicosilación avanzada, aumento de citoquinas proinflamatorias y enfermedad microvascular cerebral19. La contribución de esta última en la progresión de la EA ha sido reconocida desde hace muchos años en los estudios post morten, en los que múltiples lesiones isquémicas se han observado en las placas neuríticas típicas de la EA20. Adicionalmente un cuerpo creciente de evidencia sugiere que la IR puede jugar un rol importante en el desarrollo de la EA independientemente de los niveles de glucemia en la sangre periférica, como ha sido reportado en algunos estudios longitudinales21,22. Recientemente, Talbot y col23 mostraron una disminución de la señalización de insulina en cortes de hipocampo de pacientes fallecidos con EA, lo cual constituye la primera evidencia de IR en cerebro humano. En este estudio se muestra como luego de incubar los cortes de cerebro en diferentes concentraciones de insulina, por técnicas de inmunoprecipitación, se observa una disminución del efecto de la insulina sobre la fosforilación en diferentes etapas de la vía de señalización (IRS-1 pY, IRS-1 pS, PI3K p85α unida a IRS, Akt 1pS) en los sujetos con EA cuando se compara con sujetos sin la enfermedad.
Así, se ha propuesto a la IR como inductora de una serie de cambios en los mecanismos moleculares que dan origen a alteraciones en la síntesis y degradación de Aβ, hiperfosforilación de la proteína TAU, incremento del estrés oxidativo y producción de neuroinflamación, que en conjunto conducen a la neurodegeneración y muerte celular observada en la EA24 (Figura 1). Lo atractivo de esta hipótesis metabólica/IR cerebral es que puede explicar casi todas las alteraciones observadas en la EA, permitiendo que la responsabilidad del daño neuronal no repose exclusivamente sobre las placas neuríticas de Aβ y los ovillos de proteína TAU hiperfosforilada como tradicionalmente se ha propuesto.
Fig. 1. Esquema modificado de Son S, Shin HJ and Mook-Jung I. Insulin Resistance and Alzheimer’s Desease. In: Zimering Edited. Topics in the prevention, treatment and complications in type 2 diabetes24.
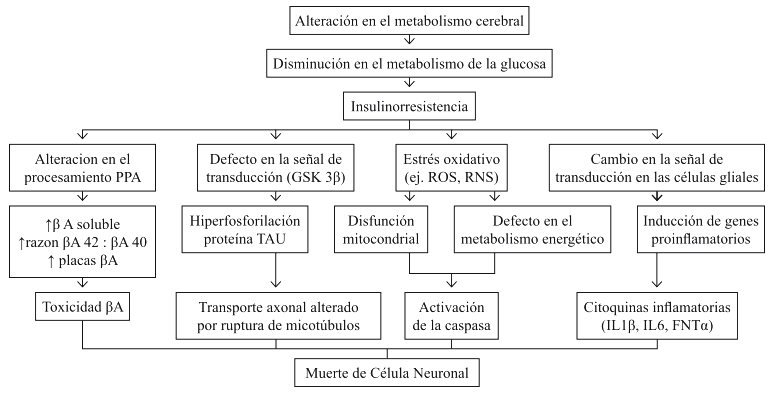
PPA: Precursor de la Proteina Amiloide. GSK 3β: Kinasa de la Glucógeno Sintetasa 3 beta. ROS: Especies Oxígeno Reactivas. RNS: Especies Nitrógeno Reactivo. βA: Beta Amiloide.
IL 1β: Interleuquina 1 beta. IL 6: Interleuquina 6. FNT α: Factor de Necrosis Tumoral alfa.
El rol de la IR como causa y consecuencia de las alteraciones observadas en la EA, ha adquirido gran interés en los últimos años. Parece existir una relación recíproca entre ambas, ya que por una parte, el concepto de que la toxicidad de Aβ causa IR está soportado por el estudio de Bomfim y col25 quienes demostraron que tanto en cultivos de neuronas de hipocampo, como la administración intraventricular en monos de oligómeros de Aβ (O Aβ), sinaptotoxina que se acumula en el cerebro de sujetos con EA desde etapas tempranas de la enfermedad, activan la vía TNFα/JNK que inducen la fosforilación SRI-pSer inhibiendo la vía fisiológica SRI-pTir. Por otra parte, el argumento opuesto en el que la IR cerebral origina alteraciones en el procesamiento y degradación de Aβ, ha sido científicamente soportada en el estudio de Ho y col26 en el que la alimentación de ratones transgénicos con modelo de EA, con una dieta alta en grasas inductora de IR mostró un incremento en la cantidad de placas neuríticas asociada a una disminución de la actividad inhibitoria de la vía fisiológica de la insulina sobre la glucógeno sintetasa 3 kinasa alfa, que ha sido relacionada con la generación de Aβ27.
DIAGNÓSTICO DE LA ENFERMEDAD DE ALZHEIMER
El diagnóstico actual de la EA es eminentemente clínico y se hace con base en los criterios especificados en el Manual Diagnóstico y Estadístico de los Trastornos Mentales (DSM-IV TR) de la Asociación Psiquiátrica Americana28, y en la Clasificación Internacional de Enfermedades (CIE-10) de la Organización Mundial de la Sa- lud29. El DSM-IV TR considera que se está frente a una demencia cuando hay: 1) deterioro de la memoria a corto y largo plazo; 2) presencia de algún trastorno cognitivo (afasia, agnosia o alguna alteración en la función de ejecución); y 3) deterioro de la actividad social y ocupacional subsecuente a los puntos 1 y 2, que no deben aparecer exclusivamente durante el curso de un estado confusional (delirium)28,29.
Más recientemente se ha considerado la inclusión de estudios adicionales a los criterios clínicos,como evaluación genética, biomarcadores y neuro-imágenes, los cuales puedan proporcionar mayor precisión diagnóstica30, aunque el diagnóstico definitivo de la EA continúa siendo el examen histopatológico post-morten. Los biomarcadores usualmente utilizados para diagnóstico de EA y para establecer la progresión de deterioro mental incipiente (DCI) a EA son los niveles en líquido cefalorraquídeo (LCR) de Aβ-42, TAU total y fosfo-TAU31,32; su sensibilidad y especificidad diagnóstica en algunos estudios es del 85%31,33, sin embargo, no son pruebas estandarizadas y para otros autores no son suficientes. Dado el amplio espectro de anormalidades que preceden o acompañan la presentación de la EA, se propone la inclusión de un panel de biomarcadores donde estén representados la mayor parte de los factores de riesgo, no obstante, el mayor desafío es la se- lección de cuales marcadores incluir de forma que costo/beneficio justifique su utilización34. Biomarcadores en sangre periférica albergan al- guna promesa como herramienta de pesquisa no invasiva35.
No hay duda del enorme progreso que se ha logrado en la detección de neurodegeneración con la aplicación de herramientas no invasivas, especialmente con el uso de neuroimágenes funcionales, las cuales han mostrado patrones de actividad cerebral aberrantes en personas de alto riesgo de EA. Entre las neuroimágenes utilizadas están: Resonancia magnética convencional (RM), Resonancia magnética funcional (RMf), Resonancia magnética con tensor de difusión (RMTD), Tomografía computarizada con emisión de positrones simple (PET) y Tomografía con emisión de positrones con 18 fluorodeoxiglucosa (FDG- PET)36.
En poblaciones a riesgo de EA como portadores de Apo E4 se ha reportado reducido metabolismo de la glucosa y/o reducida conectividad en estado de reposo en la red en modo automático (MA) del cerebro, antes de que el declive funcional sea evidente36. La red en MA, que incluye la corteza cingulada posterior y temporoparietal posterior asociadas a la región cortical del cerebro, es más activa en estado de reposo y se desactiva durante
la función cognitiva. Musen y col37 compararon el cerebro de sujetos con DM 2 con el de no diabéticos utilizando RMf y encontraron que los sujetos diabéticos tenían una reducida conectividad en estado de reposo en la red MA comparada con los controles no diabéticos, mientras que no se encontró diferencia entre los grupos de estudio en lo relativo a estructura cerebral o desempeño cognitivo. Estos resultados son concordantes con los obtenidos en sujetos portadores de apo E4, donde las alteraciones observadas en las neuroimágenes funcionales preceden al deterioro cognitivo o estructural cerebral.
El estudio de Hoogenboom y col38 utilizando RMTD mostró alteración de la integridad de la materia blanca del haz cingulado y tendencia a diferencia en los fascículos uncinado y longitudinal superior en sujetos diabéticos al compararlos con controles.
TRATAMIENTO
Cambios en el estilo de vida:Ajustes en el estilo de vida sería la forma más lógica y costo efectiva de reducir la morbi-mortalidad por EA. Recientemente se evaluó el efecto de una dieta alta en grasas y alto índice glucémico comparada con el de una dieta baja en grasas y bajo índice glucémico administrada durante 4 semanas a un grupo de 49 sujetos, un grupo de adultos saluda- bles y otros con DCI; se encontró que la dieta baja en grasas mejoró la memoria visual en ambos grupos mientras que la dieta alta en grasas incrementó Aβ en LCR de sujetos saludables, lo que sugiere que la dieta puede ser un poderoso factor ambiental que modula el riesgo de EA39. Por otra parte, un pequeño estudio evaluó el efecto del ejercicio aeróbico intenso comparado con actividades de estiramiento realizados por un grupo de 33 participantes que incluyó mujeres y hombres con DCI, se evidenció que el ejercicio aeróbico intenso tenía efecto favorable especialmente en mujeres, donde se observó mejoría en el funcionamiento cognitivo así como disminución en los niveles de Aβ en LCR40.
Farmacoterapia: El reconocimiento de las importantes funciones de la insulina en el cerebro y de la asociación de alteraciones en la vía de señalización de la insulina con la EA, ha motivado el reciente interés de evaluar los agentes terapéuticos originalmente desarrollados para tratar DM 2 como potenciales terapias en el manejo de la EA. A continuación se revisará el estado actual de la investigación de algunos de estos fármacos.
Metformina: Es poco lo que se conoce sobre el efecto de la metformina en SNC. En estudios experimentales en roedores, se observa que atraviesa la barrera hemato-encefálica y activa la AMPK en los tejidos cerebrales41, y en cultivos de células neuronales aumenta la sensibilidad a la insulina y previene las alteraciones patológicas típicas de la EA42, sin embargo, se ha reportado que la metformina aumenta la generación de Aβ en líneas celulares neuronales43. Estudios epidemiológicos44 y caso-control45 han mostrado resultados contradictorios en sujetos diabéticos con EA recibiendo metformina. Actualmente se encuentran en desarrollo estudios clínicos identificados con los números NCT00620191 y NCT01965756 con la esperanza de aclarar si la metformina puede ser de utilidad en el manejo de la EA.
Agonistas de los receptores activados por proliferadores de peroxisomas (PPARs): Los resultados con rosiglitazona no han sido uniformes, pues en un pequeño estudio clínico realizado en sujetos no diabéticos con criterios diagnósticos de EA se preservó el desempeño en las pruebas de memoria y atención en relación al placebo46, sin embargo, un estudio posterior encontró que esta mejoría estaba asociada a la presencia del alelo ApoE447. Otro estudio más reciente, reportó el efecto de la rosiglitazona como negativo respecto a las funciones cognitivas48.
Análogos del péptido similar al glucagón tipo 1 (GLP-1): Estudios experimentales con análogos de la GLP-1 han mostrado resultados favorables. Inicialmente con exenatide se observó una disminución del acúmulo de Aβ y de la toxicidad celular en modelos animales de EA49; posteriormente, en ratones transgénicos con modelo de EA, liraglutide previno alteraciones de la memoria, pérdida de las sinapsis y deterioro de la plasticidad sináptica; también redujo acúmulo de Aβ la placa neurítica e incrementó la neurogénesis en el hipocampo50, sin embargo, estos efectos no han sido demostrados en humanos.
Insulina: Los primeros reportes de estudios favorables de la utilización de la insulina sobre la función cognitiva fueron utilizando infusiones endovenosas51, sin embargo, esta vía tenía limitaciones por el riesgo de hipoglucemia y adicionalmente porque el paso de la insulina desde la periferia al SNC es limitado. Los trabajos más recientes con el uso de insulina intranasal para el tratamiento de la EA han mostrado seguridad con su uso y resultados prometedores, el más notable de ellos es el de Craft y col52 quienes evaluaron 104 pacientes con DCI o EA en los que se administró insulina intranasal diaria durante 4 meses, y en comparación con placebo, aquellos que recibieron insulina mostraron mejoría significativa de la memoria. A un subgrupo de pacientes se les realizó FDG PET con hallazgos que apoyaron los resultados clínicos, y también biomarcadores en LCR, los cuales no mostraron diferencias entre los grupos de estudio, pero el análisis exploratorio posterior mostró cambios en algunos cocientes.
Debido a estos resultados alentadores de la insulina intranasal a corto plazo, el NIH (National Institutes Health) patrocina un estudio que se está llevando a cabo en la actualidad identificado como: “Study of Nasal Insulin in the Fight Against Forgetfulness” (SNIFF) NCT01767909, donde se evaluará la eficacia de la insulina intranasal administrada a largo plazo (12 meses).
CONCLUSIONES
Varios mecanismos pueden estar involucrados en el acelerado declive cognitivo y desarrollo de EA en los sujetos con IR o DM2, sin embargo, en este escrito solo se revisaron las anormalidades metabólicas y especialmente la IR. Producto de alteraciones en la vía de señalización de la insulina se ocasiona un déficit en la utilización de la glucosa y del metabolismo energético cerebral, favoreciendo procesos inflamatorios y oxidativos que promueven el daño estructural y funcional del cerebro, que unido a la enfermedad microvascular coexistente, también favorecida por la IR, contribuyen a la expresión neuropatológica de la EA. Afortunadamente se han realizado grandes avances en el diagnóstico de la EA, en gran medida gracias al desarrollo de las neuroimágenes funcionales y de algunos biomarcadores de la enfermedad; apesar de ello, el diagnóstico de EA continúa siendo un desafío, especialmente en manos de los no especialistas en el área o de instituciones que no tengan acceso a los métodos diagnósticos más recientemente incorporados, unido al requerimiento, dada la naturaleza pro- gresiva de la EA, de largos intervalos de tiempo necesarios para demostrar los síntomas y signos de la enfermedad.
La prevención debería ser el manejo ideal de esta patología, sin embargo, el hecho de que varias funciones celulares estén comprometidas, hace pensar que esta orientación será multimodal, prestando atención a los factores de riesgo modificables. En relación a la terapia farmacológica, hay en este momento varios fármacos en evaluación, en especial los aprobados para el control de DM 2 que actúan por el mecanismo de la vía de señalización de la insulina. Sin duda, aún se requieren muchas investigaciones que permitan consolidar los hallazgos hasta ahora encontrados, que finalmente conduzcan a la prevención y tratamiento de esta incapacitante enfermedad.
REFERENCIAS BIBLIOGRÁFICAS
1.Organización Mundial de la Salud. Demencia. Abril 2016. Accesado en Junio 2016. Disponible en: www. who.int/mediacentre/factsheets/fs362/es. [ Links ]
2.Jellinger KA. The neuropathological diagnosis of Alzheimer disease. J Neural Transm Suppl 1998;53:97- 118. [ Links ]
3.Jorm AF. Risk factors for Alzheimer’s desease. In: Burn A, O’Brien J, Ames D. Editors. Dementia 3° Ed. USA. 2012.
4. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA 1997;278:1349-1356. [ Links ]
5. Peters A. The selfish brain: competition for energy resources. Am J Hum Biol 2011;23:29-34. [ Links ]
6.Rulifson EJ, Kim SK, Nusse R. Ablation of insulin- producing neurons in flies: growth and diabetic phenotypes. Science 2002;296:1118-1120. [ Links ]
7.Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links and clinical implications. Curr Alzheimer Res 2008;5:438-447.
8. Heidenreich KA, Zahniser NR, Berhanu P, Brandenburg D, Olefsky JM. Structural differences between insulin receptors in the brain and peripheral target tissues. J Biol Chem 1983;258:8527-8530. [ Links ]
9. Florant GL, Singer L, Scheurink AJ, Park CR, Richardson RD, Woods SC. Intraventricular insulin reduces food intake and body weight of marmots during the summer feeding period. Physiol Behav 1991;49:335-338. [ Links ]
10.Brant AM, Jess TJ, Milligan G, Brown CM, Gould GW. Immunological analysis of glucose transporters expressed in different regions of the rat brain and central nervous system. Biochem Biophys Res Commun 1993;192:1297-1302. [ Links ]
11.Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, Kern W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004;29:1326- 1334. [ Links ]
12.Kopf SR, Baratti CM. Effects of posttraining administration of insulin on retention of a habituation response in mice: participation of a central cholinergic mechanism. Neurobiol Learn Mem 1999;71:50-61. [ Links ]
13.Huang C, You JL, Lee CC, Hsu K . Insulin induces a novel form of postsynaptic mossy fiber long-term depression in the hippocampus. Mol Cell Neurosci 2003;24:831-841. [ Links ]
14.White MF. Insulin signaling in health and disease. Science 2003;302:1710–1711.
15.de la Monte S, Wands J. Alzheimer’s disease is type 3 diabetes – evidence reviewed. J Diabetes Sci Technol 2008;2:1101-1113.
16.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999;53:1937-1942. [ Links ]
17. Peila R, Rodriguez BL, Launer L. Type 2 diabetes, APO E gene, and the risk for dementia and related pathologies: The Honolulu- Asia Aging Study. Diabetes 2002;51:1256-1262. [ Links ]
18.Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 2010;67:505-512.
19.Whitmer RA. Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep 2007;7:373–380.
20.Etiene D, Kraft J, Ganju N, Gomez-Isla T, Gemelli B, Hyman BT, Hedly-White ET, Wands JR, De la Monte SM. Cerebrovascular pathology contributes to the heterogeneity of Alzheimer’s disease. J Alzheimers Dis 1998;1:119-134.
21.Rönnemaa E, Zethelius B, Sundelöf J, Sundström J, Degerman-Gunnarsson M, Berne C, Lannfelt L, Kilander L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008;71:1065-1071. [ Links ]
22.Schrijvers E, Witteman J, Sijbrands E, Hofman A, Koudstaal P, Breteler M. Insulin metabolism and the risk of Alzheimer ́s disease: the Rotterdam Study. Neurology 2010;75:1982-1987. [ Links ]
23.Talbot K, Wang HY, Kazi H, Han HY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline J. Clin. Invest 2012;122:1316-1338.
24. Son S, Shin HJ, Mook-Jung I. Chapter 3: Insulin resistance and Alzheimer’s desease. In: Zimering. Topics in the prevention, treatment and complications in type 2 diabetes. Edited by Mark B. 2011. Accessed May 2016. Available in: http://www.intechopen.com/books/topics- in-the-prevention-treatment-and-complications-of-type-2-diabetes/insulin-resistance-and-alzheimer-s-disease
25 Bomfim TR, Forny-Germano L, Sathler LB, Brito- Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest 2012;122:1339-1353.
26.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A. Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 2004;18:902-904.
27.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid beta peptides. Nature 2003;423:435-439
28. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939-944.
29.Guía de Clasificación de los Trastornos Mentales y del Comportamiento CIE-10: CDI-10. Editorial Médica Panamericana, 2000. [ Links ]
30.DeKosky ST, Carrillo MC, Phelp C, Knopman D, Petersen RC, Frank R, Schenk D, Masterman D, Siemers ER, Cedarbaum JM, Gold M, Miller DS, Morimoto BH, Khachaturian AS, Mohs RC. Revision of the criteria for Alzheimer’s disease: a symposium. Alzheimers Dement 2011;7:e1-12.
31.Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter WZ, Weiner MW, Jack CR Jr, Jagust W, Toga AW, Lee VM, Shaw LM. Alzheimer’s Disease Neuroimaging Initiative. Update on the biomarker core of the Alzheimer’s disease neuroimaging initiative subjects. Alzheimers Dement 2010;6:228-230.
32. Blennow K, Zetterberg H. Cerebrospinal fluid biomarkers for Alzheimer’s disease. J Alzheimers Dis 2009;18:413-417.
33.Monge-Argilés JA, Sánchez-Payá J, Muñoz-Ruiz C, Pampliega-Pérez A, Montoya-Gutiérrez J, Leiva- Santana C. Biomarkers in the cerebrospinal fluid of patients with mild cognitive impairment: a meta- analysis of their predictive capacity for the diagnosis of Alzheimer’s disease. Rev Neurol 2010;50:193-200.
34. Mattsson N, Blennow K, Zetterberg H. CSF biomarkers: pinpointing Alzheimer pathogenesis. Ann NY Acad Sci 2009;1180:28-35. [ Links ]
35.Schneider P, Hampel H, Buerger K. Biological marker candidates of Alzheimer’s disease in blood, plasma, and serum. CNS Neurosci Ther 2009;15:358-374.
36.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for lateonset Alzheimer’s dementia. Proc Natl Acad Sci U S A 2004;101:284-289.
37.Musen G, Jacobson AM, Bolo NR, Simonson DC, Shenton ME, McCartney RL, Flores VL, Hoogenboom, WS. Resting-state brain functional connectivity is altered in type 2 diabetes. Diabetes 2012;61:2375-2379. [ Links ]
38.Hoogenboom WS, Marder TJ, Flores VL, Huisman S, Eaton HP, Schneiderman JS, Bolo NR Simonson DC, Jacobson AM, Kubicki M, Shenton ME, Musen
G. Cerebral white matter integrity and resting-state functional connectivity in middle-aged patients with type 2 diabetes. Diabetes 2014;63:728-738.
39.Bayer-Carter JL, Green PS, Montine TJ, VanFossen B, Baker LD, Watson GS, Bonner LM, Callaghan M, Leverenz JB, Walter BK, Tsai E, Plymate SR, Postupna N, Wilkinson CW, Zhang J, Lampe J, Kahn SE, Craft S. Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Arch Neurol 2011;68:743-752. [ Links ]
40.Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, McTiernan A, Plymate SR, Fishel MA, Watson GS, Cholerton BA, Duncan GE, Mehta PD, Craft S. Effects of aerobic exercise on mild cognitive impairment. A controlled study. Arch Neurol 2010;67:71-79. [ Links ]
41.Łabuzek K, Suchy D, Gabryel B, Bielecka A, Liber S, Okopien B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep 2010;62:956-965. [ Links ]
42.Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011;60:910-920.
43.Chen Y, Zhou K, Wang R, Liu Y, Kwak Y-D, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Lu Z, Xu H, Li F-F. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA 2009;106:3907-3912.
44.Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis 2011;24:485-493. [ Links ]
45.Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case-control study. J Am Geriatr Soc 2012;60:916-921.
46.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, Fishel MA, Kulstad JJ, Green PS, Cook DG, Kahn SE, Keeling ML, Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry 2005;13:950-958. [ Links ]
47.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD; Rosiglitazone in Alzheimer’s Disease Study Group. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J 2006;6:246-254.
48.Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M,Craft S, Landreth G, Linnamägi Ü, Sawchak S. Rosiglitazone monotherapy in mild-to-moderate alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 2010;30:131-146.
49.Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis 2010;19:1205-1219.
50.McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci 2011;31:6587-6594.
51. Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 2001;74:270-280. [ Links ]
52.Craft S, Baker LD, Montine TJ, Satoshi M,Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 2012;69:29-38. [ Links ]
