










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Puericultura y Pediatría
versión impresa ISSN 0004-0649
Arch Venez Puer Ped vol.76 no.4 Caracas dic. 2013
Epilepsia y enfermedades neurocutáneas. Abordaje en neuropediatría.
María Gabriela Jiménez Méndez(1), Freda Hernández de París(2),Laura S Calzadilla(3), Gabriela del Valle Ríos(3), Fátima Correia(3)
(1) Pediatra. Residente de tercer año de Neurología Pediátrica. Universidad del Zulia. Hospital Universitario de Maracaibo
(2) Neuropediatra, Jefe del Servicio de Neuropediatría Hospital Universitario de Maracaibo
(3) Neuropediatra. Adjuntos del Servicio de Neuropediatría, Hospital Universitario de Maracaibo
Segundo Premio del LIX Congreso Nacional de Pediatría 2013
Autor corresponsal: María Gabriela Jiménez Méndez 0414-617.0727, 0261-743.5937 marigabijimenez@hotmail.com, mariagabrielajimenez@hotmail.com
RESUMEN
Introducción: La epilepsia es generalmente la primera manifestación que obliga a la consulta con neurología pediátrica pasando por alto las lesiones de la piel encontradas en la mayoría de las Enfermedades Neurocutáneas (EN), las cuales son trastornos determinados genéticamente que afectan selectivamente a órganos y tejidos derivados del ectodermo embrionario. Objetivo: Conocer las características de la epilepsia en los pacientes pediátricos con EN que asisten a la consulta de neuropediatría del Hospital Universitario de Maracaibo entre 2010 y 2013 Método: estudio observacional tipo serie de casos Resultados: Veintiún pacientes cumplieron los criterios de EN, 17 presentaron crisis epilépticas, 9 niñas y 8 niños. La Esclerosis Tuberosa (ET) fue la causa más frecuente seguida del síndrome de Sturge Weber. Se encontró significación estadística al correlacionar el inicio precoz de las crisis convulsivas antes de los 24 meses de edad y el diagnóstico de ET. Las crisis focales fueron más frecuentes que las generalizadas. El valproato y la oxcarbazepina en monoterapia o combinados son los medicamentos de elección. El retardo mental fue la comorbilidad más frecuentemente encontrada. Conclusiones: La ET fue la principal EN asociada a epilepsia con un mayor riesgo de inicio de crisis antes de los 24 meses, seguida del Síndrome de Sturge Weber. No existe una asociación significativa entre el sexo, el grupo etario y el riesgo de epilepsia. La severidad variable de las crisis amerita en muchos casos politerapia farmacológica para su control.
Palabras Clave: Epilepsia, Enfermedades Neurocutáneas, niños
Epilepsy and neurocutaneous diseases. Approach in neuropediatrícs.
SUMMARY
Epilepsy is usually the first sign that requires neurological consultation often overlooking the visible skin lesions so common in most neurocutaneous diseases, genetically determined disorders that selectively affect organs and tissues derived from the embryonic ectoderm Objective: To determine the characteristics of epilepsy in pediatric patients with neurocutaneous diseases attending the neuropaediatric clinic at the University Hospital of Maracaibo between 2010 and 2013 Methods: Observational case series Results: Twenty one patients met the criteria for neurocutaneous disorders. Epilepsy was seen in 17 patients, 9 were female and 8 were male. Tuberous sclerosis was the most frequent cause, followed by Sturge Weber syndrome. Statistical significance was found for the association of early onset epileptic crisis before 24 months of age and tuberous sclerosis. Focal seizures were more frequent than generalized seizures. Valproate and oxcarbazepine as monotherapy or in combination are the drugs of choice. Mental retardation was the most frequent comorbidity found Conclusions: Tuberous sclerosis was the major neurocutaneous disease associated with epilepsy with an increased risk of onset of crisis before 24 months of age, followed by Sturge Weber syndrome. There is no significant association between gender and age group and risk of epilepsy. Variable severity of epileptic crises requires polytherapy for adequate control in many cases.
Key words: Epilepsy, Neurocutaneous Diseases, children
Recibido: 15-09-2013 Aceptado: 20-11-2013
INTRODUCCIÓN
La asociación de epilepsia con otras disfunciones neurológicas como retardo mental, alteraciones de la conducta, autismo, demora en la adquisición de habilidades motoras, plantean un abanico de posibilidades diagnósticas, las cuales sumadas a otras manifestaciones sistémicas y dermatológicas obligan a considerar una Enfermedad Neurocutánea (EN) como causa subyacente.
La relación inequívoca de algunas enfermedades neurológicas con alteraciones cutáneas siempre ha atraído la atención de los médicos. Muchas han sido reconocidas como condiciones relativamente raras, pero en conjunto, representan una proporción significativa de los trastornos neurológicos, especialmente en los niños. Sus manifestaciones clínicas son polimórficas o heterogéneas y de expresividad variable, involucrando muchos otros órganos o sistemas, además de la piel y el sistema nervioso (1).
Las EN son trastornos determinados genéticamente, que afectan selectivamente a órganos y tejidos derivados del ectodermo embrionario, pudiendo comprometer el encéfalo, la médula espinal y los nervios periféricos, así como otros órganos blancos y el sistema óseo (2,3). La epilepsia por lo general es la primera manifestación que obliga a la consulta
con un neurólogo pasando muchas veces por alto las lesiones visibles de la piel u otras manifestaciones. No en todas las EN la epilepsia es la principal manifestación pero sigue siendo la alarma para ser derivado a un servicio de neurología. Son muchas las EN que cursan con convulsiones; es así como en la Esclerosis Tuberosa (ET) es la manifestación más importante. Se estima que 80 a 90% de las personas con ET tendrán convulsiones de inicio frecuente en la niñez; y que desafortunadamente, en algunos casos resulta intratable con medicamentos (4). Son particularmente frecuentes los espasmos infantiles en menores de un año (5). En niños mayores y adultos se desarrollan habitualmente crisis focales simples y/o complejas, con menos frecuencia crisis tónico-clónicas generalizadas, atónicas, tónicas, mioclónicas o ausencias atípicas. Es común la combinación de dos o más tipos de crisis pudiendo conformar epilepsias tipo Lennox-Gastaut (6).
Las EN están asociadas a otras manifestaciones neurológicas. Aproximadamente un tercio de los pacientes tienen retardo mental variable, generalmente leve, cefaleas, trastornos del lenguaje, trastornos adaptativos, algunos de ellos cursan con una predisposición a desarrollar tumores gliales, así mismo manifestaciones extraneurológicas y sistémicas entre las que destacan las alteraciones endocrinológicas: pubertad precoz, acromegalia, enfermedad de Addison, hiperparatiroidismo, ginecomastia y desarrollo de feocromocitoma suprarrenal, alteraciones esqueléticas como la xifoescoliosis por displasia vertebral (1). De allí la importancia de que todo paciente una vez diagnosticado sea valorado por un equipo multidisciplinario con el seguimiento oportuno de las diferentes comorbilidades (3).
La Neurofibromatosis (NF) tipo I o Neurofibromatosis de Von Recklinghausen es la EN más frecuente, en la cual la epilepsia se presenta en menos del 3,5% de la población afecta (7). En la mayoría de los casos se obtiene un control terapéutico adecuado con la medicación antiepiléptica habitual (8). En el Síndrome de Sturge Weber (SSW) también se reporta una alta asociación con crisis epilépticas, las cuales se presentan en el 55% a 90%, siendo más común en pacientes con hemangiomas faciales bilaterales donde el 75% inician en el primer año de vida como crisis focales (9,10).
En otras EN menos frecuentes como la Incontinencia Pigmenti (IP), las crisis epilépticas están presentes en un 12% de los casos, pudiendo ser focales o generalizadas, sintomáticas, asociadas a retraso mental (11). Actualmente la antes mal llamada hipomelanosis de Ito (HI) no se considera una única enfermedad, sino el resultado de diferentes mosaicismos cromosómicos por una mutación postzigótica y por lo tanto no trasmisible, por lo que se ha propuesto los términos de mosaicismo pigmetario (MP), hipomelanosis a lo largo de las líneas de Blaschko, incontinencia pigmentaria acromática o displasia pigmetaria, de los que no existen criterios diagnósticos establecidos, sino que cualquier paciente con una clínica cutánea consistente en hipopigmentación a lo largo de las líneas de Blaschko y afectación extracutánea se individualiza esta entidad como una EN (12), se asocia a epilepsia en la mitad de los casos, siendo el retardo mental su manifestación principal (13). La enfermedad de Von Hippellindau, la melanosis Neurocutánea, la Ataxia-telangiectasia, el síndrome de Proteus y el de Klippel–Trenaunay son menos frecuentes y con menor incidencia de epilepsia (1).
En las últimas décadas los estudios de genética molecular han permitido avances considerables en la comprensión de las causas, mecanismos y clasificación de las EN. Los nuevos descubrimientos continúan desafiando conceptos consagrados en el tiempo para generar nuevos como los relacionados al mosaicismo somático. El estudio de las EN también ha permitido una mejor comprensión de algunas de las causas y mecanismos de formación de tumores así como de muchos aspectos de la oncogénesis, tema principal de las investigaciones actuales y futuras, con implicaciones importantes para la salud pública y práctica médica (1,2).
El gran aporte en el desarrollo de la genética molecular y de las neuroimágenes ha permitido encontrar una visión general de la complejidad de éste grupo de condiciones. El papel del clínico consiste en detectar oportunamente y a edad precoz la asociación de los diferentes signos y síntomas neurológicos, incluyendo un examen minucioso de la piel para establecer las posibilidades diagnósticas de las EN.
En Venezuela son pocos los reportes de epilepsia y EN, más como reporte de casos aislados (14,15). El objetivo de este estudio es conocer las características y comorbilidades de la epilepsia en los pacientes con EN en la población pediátrica que asiste a la consulta de neurología pediátrica del Hospital Universitario de Maracaibo.
MÉTODOS
Se realizó un estudio observacional tipo serie de casos, mediante registro consecutivo de los pacientes pediátricos de ambos sexos, entre 0 y 15 años, con criterios clínicos de EN y epilepsia que consultaron al servicio de neuropediatría del Hospital Universitario de Maracaibo entre Enero 2010 y Enero 2013. Estos fueron analizados según aspectos epidemiológicos, edad de diagnóstico e inicio de crisis, motivo de consulta, tipo de crisis epiléptica, hallazgos electroencefalográficos y de neuroimágenes, asociación a trastornos conductuales, severidad de compromiso intelectual y manifestaciones dermatológicas, tipo de fármaco antiepiléptico (FAE) y evolución clínica. Los padres, niños y jóvenes fueron informados acerca de la naturaleza y propósito del estudio, obteniéndose el consentimiento informado por escrito de sus representantes en acuerdo con la Declaración de Helsinki II.
Se aplicó para el análisis descriptivo de las variables cuantitativas medidas de tendencia central y dispersión. La asociación estadística se determinó mediante la prueba del Chi cuadrado, y/o el test Exacto de Fischer para las variables cualitativas, tomando un valor de p <0,05 como estadísticamente significativo. Se determinó la razón de proporcionalidad (Odds Ratio) para establecer la fuerza epidemiológica de relación entre las variables cualitativas principales.
RESULTADOS
De los pacientes evaluados, 21 cumplieron criterios diagnósticos para EN, de los cuales 17 cursaban con crisis epilépticas, excluyendo 4 pacientes: 2 ataxia telangiectasia, 1 melanosis neurocutánea, 1 NF tipo 1 y tipo 2 respectivamente, lo cuales no presentaron crisis epilépticas durante el estudio.
En los casos estudiados con epilepsia, la ET ocupa el primer lugar, seguido por el SSW, y en igual número de casos en la antes llamada HI, y síndrome de PHACES acrónimo inglés utilizado para describir un síndrome caracterizado por la asociación de malformaciones de fosa posterior cerebral, grandes hemangiomas faciales, anomalías anatómicas de las arterias cerebrales, coartación aórtica, otras anomalías cardiacas, y anomalías oculares (Tabla 1).
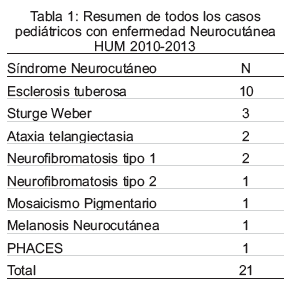
El promedio de edad fue de 5,3 ± 4,8 años, con un rango entre 1 mes y 14 años, sin predominio significativo en ningún grupo etario, con similar frecuencia en lactantes, pre escolares y escolares, y en menor porcentaje en adolescentes.
En cuanto a la distribución de epilépticos con EN según el género, 9 correspondieron al femenino y 8 al masculino (Tabla 2).
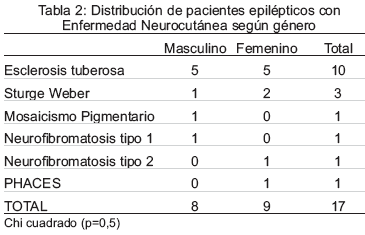
Dos pacientes tenían antecedentes de consanguinidad y 5 tenían familiares en primer grado con la misma patología neurocutánea. Las crisis epilépticas fueron el principal motivo de consulta en 16 casos, 2 de ellos debutaron con espasmos infantiles. Un sólo paciente consultó por parálisis facial periférica e hipoacusia presentando crisis convulsivas al año del diagnóstico de NF tipo 2.
La edad de inicio de las crisis epilépticas fue: 10 antes del año de edad, 4 entre los 2 y 5 años, y los 3 restantes, posterior a los 5 años. En la correlación entre la edad de inicio y las diferentes EN, 8 pacientes con ET iniciaron las crisis epilépticas de forma precoz antes de los 24 meses de edad constituyendo este un hallazgo estadísticamente significativo (p<0,05), con OR 2,8. (Tabla 3).
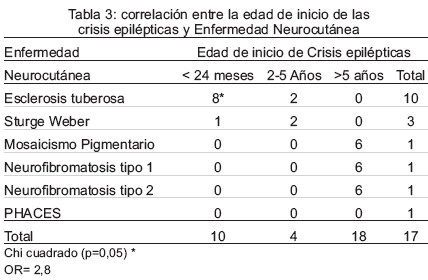
Las manifestaciones neurológicas no se presentaron de forma aislada, es decir, un mismo paciente cursaba con varias condiciones neurológicas de forma simultánea, siendo frecuente la asociación de retardo mental con demora en la adquisición de habilidades motoras. Las comorbilidades más frecuentemente asociadas a la epilepsia se especifican en la Tabla 4.
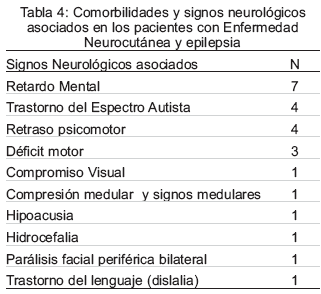
De los 17 pacientes epilépticos, 9 cursaron con crisis focales; de éstos, 4 sin compromiso del estado de conciencia (según la clasificación ILAE 2010) (16) antes crisis focales simples, y 5 tuvieron crisis focales discognitivas (antes crisis focales complejas). Los 2 pacientes que cursaron con crisis generalizadas, estas fueron tónico-clónicas (las antes llamadas gran mal). Ninguno cursó con crisis de ausencia o mioclónicas. Las crisis reflejas (n=1), crisis sutiles (n=1) y los espasmos infantiles (n=1) se encontraron en igual proporción. Se observaron 3 pacientes con ET que cursaron con síndromes electroclínicos con crisis mixtas tipo Lennox-Gastaut. La epilepsia se inició en etapas muy tempranas como un Síndrome de West ó espasmos infantiles. Un paciente debutó con crisis sutiles en el periodo neonatal y 1 paciente con crisis epilépticas reflejas (tónico clónicas) desencadenadas por estímulos auditivos diagnosticado con HI.
No existe un patrón electroencefalográfico característico en ningún caso particular de EN. Todos los electroencefalogramas de pacientes epilépticos con EN mostraron alteraciones: 12 patrón paroxístico focal, 3 con patrón hipsarrítmico, 2 patrón focal secundariamente generalizado.
La Resonancia Magnética Cerebral (RMC) se realizó en 15 de los pacientes epilépticos, dos de ellos lactantes menores de 1 y 2 meses de edad (pacientes 9 y 15 de la Tabla 5). No se logró realizar la RMC en 2 pacientes epilépticos (uno por muerte y otro SSW diferida hasta los 6 meses de edad). Catorce pacientes mostraron lesiones estructurales en correspondencia a su EN excepto 1 paciente con HI que mostró leucomalacia parasagital. Un paciente con diagnóstico de ET presentó hidrocefalia obstructiva secundaria a astrocitoma gigante (Tabla 5). Algunas características clínicas y los estudios de RMC de los pacientes 1, 14 y 17 de la Tabla 5 se pueden observar en las Figuras 1-3.
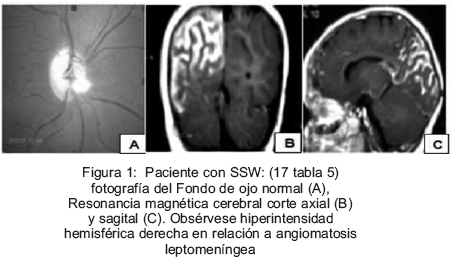

Se consideró como epilepsia de difícil control a aquella que requirió más de un fármaco para su manejo. Se observó que 8 de los pacientes tuvieron este comportamiento. En relación a la terapia anticonvulsiva, 5 pacientes recibieron monoterapia con ácido valproico (VPA), 4 con oxcarbazepina (OXC), 8 recibieron terapia combinada con dos y tres FAE. Tres pacientes han cursado con estatus convulsivos durante su evolución. Un sólo paciente con ET y síndrome de West ameritó tratamiento con esteroides. En ningún caso se ha recurrido al uso de dieta cetogénica para manejo de las crisis epilépticas, u otras terapias alternativas como estimulación del nervio vago o neurocirugía (Tabla 5).
Cuatro pacientes fallecieron durante el período del estudio: 2 ET, uno por hidrocefalia obstructiva por un Astrocitoma gigante, y el otro por una complicación cardíaca derivada de un rabdomioma cardíaco); 1 con SSW por una neumonía complicada y 1 con PHACE al mes de vida por una hemorragia cerebral masiva.
DISCUSIÓN
En la serie presentada, los tres tipos más comunes de EN fueron similares a los reportados en la literatura, diferenciándose en el orden de frecuencia ya que el primer lugar lo ocupa la ET, seguida por el SSW y en tercer lugar la NF1 y NF2, mientras que ésta última es la más frecuente en la mayoría de los reportes de casos de EN (3,8). Las formas de presentación clínica dermatológica y neurológica no presentaron diferencias significativas con respecto a estudios publicados (9,17). Se reporta una mayor asociación de epilepsia comparada con estudios similares como el de Durón R y colaboradores (3), quienes reportaron una menor asociación de casos de EN y epilepsia. Esta diferencia podría deberse a que en su serie se reporta un mayor numero casos de NF y otras EN con menor incidencia de epilepsia tales como: síndrome de Klippel Trenaunay, melanosis cutáneas, entre otras, en pacientes con un mayor rango de edad y recogidos en un lapso de 7 años versus los 3 años de éste estudio y con un mayor rango de edad.
No se registró una asociación estadísticamente significativa en relación al género y a la edad lo que coincide con la mayoría de los reportes internacionales (2). Igualmente, la baja proporción de pacientes epilépticos con antecedentes familiares de la misma EN coincide con lo reportado en dichos estudios. La edad de inicio de las crisis mostró una asociación estadísticamente significativa con el tipo de EN, encontrándose que la ET tuvo un riesgo casi 3 veces mayor de presentar crisis convulsivas antes de los 24 meses de edad. Esta EN es la responsable de la mayoría de los casos de epilepsia en comparación con el resto de las EN, en cuyo caso las crisis epilépticas se presentan más tardíamente, después de los 5 años.
En series de casos locales de pacientes pediátricos con ET se reportan hasta un 56% en asociación con epilepsia (14,15), y a pesar que las crisis epilépticas no forman parte de los criterios diagnósticos, son el motivo más frecuente de consulta (18).
El SSW constituye la segunda causa en esta serie de pacientes. Montes C y colaboradores (10) encontraron que las crisis epilépticas son el trastorno neurológico asociado más común, afectando la mayor parte de los casos (92%). La edad promedio en el inicio de las crisis fue de 1.4 años, en su mayoría de tipo parcial con o sin generalización secundaria. Otra serie de casos en México reportó que en un grupo de 20 pacientes, el 100% presentaba retraso mental, crisis generalizadas en su mayoría y especialmente síndrome de West en un 25%, otro tanto con crisis parciales (19). En 1998, Díaz C y colaboradores (20) reportaron un grupo de 30 pacientes con SSW, de los cuales el 83% presentaba crisis epilépticas, de predominio parcial, dos de ellos con debut en estatus epiléptico y 5 de ellos con epilepsia de difícil control. A dos de estos se le realizó hemisferectomía logrando el control total de las crisis. En aspectos de manejo, se observó que el fármaco de primera línea fue el VPA en más de la mitad de los pacientes, seguido de otros como el fenobarbital y el clobazam (CLB), lo cual se asemeja al manejo utilizado en el presente estudio.
Un caso de la antes llamada HI, actualmente mosaicismo pigmentario (MP), cursó con epilepsia refleja desencadenada ante estímulos auditivos y retardo mental moderado a severo, siendo ésta última la manifestación neurológica más frecuentemente descrita en ésta entidad. Osborn coincide con otros estudios en pacientes con MP (21-23) describiendo que el 50% a 75% presentan lesiones en el sistema nervioso central; retraso mental en más del 60%, crisis convulsivas refractarias al tratamiento, comportamiento autista en el 10% de los casos. El único paciente con MP de ésta serie, a pesar de cursar con retardo mental y crisis epilépticas reflejas con buena respuesta al tratamiento con OXC, no presentaba las alteraciones imagenológicas más frecuentemente descritas en ésta entidad, tales como alteraciones en la laminación cortical, heterotopias de la sustancia gris y atrofia difusa (12) mostrando únicamente leucomalacia parasagital sin ningún otro hallazgo patológico.
A pesar de ser la NF la EN más frecuente no fue alta su asociación a epilepsia en éste estudio. La prevalencia de epilepsia en pacientes con NF se sitúa entre el 3 y el 7,3% (24). Aunque la frecuencia es claramente superior a la observada en la población general, indudablemente en muchos pacientes con NF-I, la presencia de convulsiones tan sólo pone de manifiesto una mera coincidencia de dos procesos frecuentes. Al igual que ocurre con otros trastornos neurocutáneos, se han descrito diferentes tipos de convulsiones, incluidos los espasmos infantiles (25). Los 2 pacientes con NF tipo 1 y 2 cursaron con epilepsia con características similares. El primer caso presentó una afectación motora importante (tetraplejía espástica) y cursaba con crisis convulsivas recurrentes que respondieron favorablemente al VPA. El segundo caso presentó crisis focales de breve duración y sin compromiso de la conciencia con una excelente respuesta a la OXC, lo que coincide con lo descrito en la literatura en la cual se describe que en la mayor parte de los casos se obtiene un control terapéutico adecuado con la medicación antiepiléptica habitual.
En la presente serie solo se reportó un caso de PHACE, en el que se presentaron anomalías del esternón adicionalmente a las descritas tradicionalmente, lo cual le confiere la denominación de PHACES. A pesar que muchos casos que describe la literatura evolucionan favorablemente la paciente falleció antes de los 3 meses de edad.
Finalmente, es de gran interés para el clínico que se enfrenta a cuadros convulsivos recordar la asociación embriológica neuroectodermal y sus derivados cerebro-piel, de manera que la presencia de crisis convulsivas y cualquier tipo de lesión en piel y faneras, deben hacer sospechar una EN o Facomatosis. Las EN requieren tratamiento y seguimiento multidisciplinario de por vida, incluyendo especialmente a pediatras, dermatólogos, genetistas, neurólogos, neurocirujanos, ortopedas, oftalmólogos, endocrinólogos y cirujanos vasculares entre otros. Debe vigilarse a los pacientes por complicaciones oncológicas, esperadas en alrededor de 10% de los casos. La valoración genética es fundamental en todos los pacientes con EN, lo cual constituye una de las limitaciones de éste estudio, debido a que, aún cuando todos los pacientes fueron referidos para la respectiva evaluación genética, esta no se cumplió en la totalidad de los pacientes.
REFERENCIAS
1. Rugieri M, Pascual-Castroviejo I., Di-Rocco C, eds. Neurocutaneous Disorders, phakomatoses and hamatoneoplastic syndromes. Springer Wien; New York, USA 2008. 1070 p. [ Links ]
2. Rufo-Campos M, Rufo-Muñoz M. Trastornos neurocutáneos. Pediatr Integral 2003; 7 (8):603-613. [ Links ]
3. Durón R, Lizardo G, López E, Morales S, Hesse H, Molina L, et al. Síndromes neurocutáneos en la consulta neurológica. Serie de casos. Rev Med Hondur 2009; 77(4):172-176. [ Links ]
4. Henske E, Scheithauer B, Short M, Wollmann R, Nahmias J, Van Slegtenhorst M, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet 1996; 59: 400-406. [ Links ]
5. Asano E, Juhasz C, Shah A, Muzik O, Chugani D, Shah, J, et al. Origin and propagation of epileptic spams delineated on electrocorticography. Epilepsia 2005;46:1086-1097. [ Links ]
6. Gomez MR. Phenotypes of the Tuberous Sclerosis Complex with a revision of diagnostic criteria. En: Johnson W.G., & Gomez M.R. Eds. Tuberous sclerosis and allied disorders: Clinical, cellular, and molecular studies. New York: New York Academy of Science 1991. 615: 1-7. [ Links ]
7. Pascual-Castroviejo I. Neurofibromatosis tipo I (NF-I): peculiaridades y complicaciones. Rev Neurol 1996; 24: 1051-1055. [ Links ]
8. Karnes P. Neurofibromatosis: a common neurocutaneous disorder. ClinProc 1998; 73:1071-1076. [ Links ]
9. Garzon M, Huang J, Enjolras O, Frieden I. Vascular malformations. Part II: associated syndromes. J Am Acad Dermatol 2007;56 (4):541-564. [ Links ]
10. Montes C, Barragán E, Legido S. Características de la epilepsia en pacientes con síndrome de Sturge-Weber. Serie de casos del Hospital Infantil de México. Rev Med Hondur 2010; 78 (4):169-224. [ Links ]
11. Matelzonas T, Ruvertoni M, Reyno S, Pinchak M. Incontinentia pigmenti. Presentación neonatal. A propósito de un caso clínico. Arch Pediatr Urug 2010; 81(1): 23-29. [ Links ]
12. Ruggieri M, Pavone L. Hypomelanosis de Ito: Clinical syndrome or just phenotype?. J Child Neurol 2000; 15: 635- 644. [ Links ]
13. Assogba K, Ferlazzo E, Striano P, Calarese T. Heterogeneous seizure manifestations in Hypomelanosis of Ito: report of four new cases and review of the literature. Neurol Sci 2010; 31: 9-16. [ Links ]
14. Jiménez M, Lacruz-Rengel M, Camarata F, Girard Y, Jaimes E. Serie de casos de Esclerosis Tuberosa en la población infantil del Instituto Autónomo Hospital Universitario de los Andes, Mérida – Venezuela. Pediátr Panamá 2011; 40 (2): 20-24. [ Links ]
15. Jiménez M, Lacruz-Rengel M, Camarata F. Epilepsia en niños con esclerosis tuberosa experiencia en el Instituto Autónomo Hospital de los Andes. Mérida 2005-2011. Arch Venez Puer Ped 2011; 74 (3): 112-117. [ Links ]
16. Berg A, Berkovic S, Brodie M, Buchhalter J, Cross J, Van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commissions on classification and terminology, 2005-2009. Epilepsia 2010; 51(4): 676-685. [ Links ]
17. Rodríguez-Díaz E, de Unamuno P. El síndrome de Sturge- Weber. MedClin 1993; 101:18-19. [ Links ]
18. Holmes G, Staftrom C. Tuberous Sclerosis Complex and Epilepsy: recent development and future challenges. Epilepsia 2007;48:617-630. [ Links ]
19. Ruben E, Francisco L. Perfil clínico- epidemiológico del Síndrome de Sturge Weber. Rev Mex Neuroci 2008; 9(2):106- 118. [ Links ]
20. Díaz C, García M. Síndrome de Sturge Weber. Acta Pediatr Mex 1998;19 (4):180-182. [ Links ]
21. Osborn A. Neurorradiología Diagnóstica. Trastorno de la histogénesis: Síndromes neurocutáneos. Mosby eds; Madrid, 1996. 110 p. [ Links ]
22. Swaiman K. Neurología Pediátrica. Principios y prácticas. Síndromes neurocutáneos: facomatosis y trastornos relacionados. 2da edición. Mosby - Doyma eds; Madrid, 1996. 1084 p. [ Links ]
23. Kulkarni M, Kumar C, Venkataramana V, Reddy E. Hypomelanosis of Ito. Indian Pediatr. 1996;33 (3):243-245. [ Links ]
24. Pascual-Castroviejo I. Complications of neurofibromatosis type I in a series of 197 children. En Fukujama Y, Suzuki Y, Kamoshita S, Casaer P, eds Fetal and perinatal neurology. Basel Karger; 1992. pp 162-173. [ Links ]
25. King A, Upadhyaya M, Penney C, Doshi R. A case of Miller Dieker syndrome in a family with neurofibromatosis type I. Acta Neuropathol 2000; 99:425-427. [ Links ]
