










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkActa Odontológica Venezolana
versión impresa ISSN 0001-6365
Acta odontol. venez v.40 n.2 Caracas jun. 2002
SÍNDROME DE ECTRODACTILIA-DISPLASIA ECTODÉRMICA-HENDIDURA ( EEC. )
Revisión de la Literatura. Reporte de un caso.
Cristina Umérez. Odontopediatra. U.C.V. Miembro de la Sociedad Venezolana de Odontopediatria.
Rafael Darío Sosa. Cirujano Bucal. Profesor Asistente de la Cátedra de Cirugía Estomatológica
Facultad de Odontología U.C.V. Miembro de las Sociedades de Patología Bucal y Cirugía Buco-Maxilofacial Venezolanas.
Dr. Venancio Simosa.
Médico Genetista del Instituto de Medicina Experimental de la Facultad de Medicina U.C.V y del Hospital José Gregorio Hernández de Magallanes de Catia.
Profesor colaborador del postgrado de Odontopediatría de la Facultad de Odontología U.C.V.
RESUMEN
Este síndrome se caracteriza por la presencia de la triada Ectrodactilia-Displasia Ectodérmica-Hendidura (EEC). Esta asociación muchas veces es confundida con otro tipo de identidades quedando así erróneamente diagnosticado el paciente, por lo cual se plantean variados diagnósticos diferenciales del mismo. El siguiente trabajo tiene como objetivo la presentación de un caso clínico de un paciente masculino de 5 años de edad con el diagnóstico presuntivo de Displasia Condroectodérmica y el cual cursaba con hendidura labio-palatina completa bilateral asociada a discromía del cabello, ectrodactilia de manos y pies, polidactilia post-axial, trastornos dentarios y obstrucción de ambos ductos lagrimales. Para tal efecto, se realizó un estudio genético, examen clínico, radiografía panorámica y de tórax, evaluación oftalmológica y neurológica. De estos exámenes se pudo concluir que en base a las características genéticas y clínicas representaba un caso de Síndrome EEC. Estos resultados sugieren que el diagnóstico de este tipo de identidades clínico-patológicas debe realizarse conjuntamente por un grupo de especialistas mèdico-odontologicos, para de esta forma establecer un plan de tratamiento adecuado para el paciente. Finalmente se describe en detalle el diagnóstico y plan de tratamiento dental instaurado en el paciente.
Palabras Claves: Síndrome, Displasia, Hendidura.
ABSTRAC
Ectrodactyly-Ectodermal dysplasia-Fissure (EEC) is a condition characterized for the presence of this triad. Many times this association is confused with other type of entities being the patient misdiagnosed, reason why are varied differential diagnoses established.The present work is a clinical case of a 5 year old male patient with the presumtive diagnosis of Chondroectodermal Dysplasia which courses with complete bilateral lip palatal fissure associated with hair dyschromia, feet and hands ectrodactyly,post-axial polydactyly, dental alterations and obstruction of both lacrimal ducts. A genetic study, clinical exam, panoramic and torax X rays, and ophtalmological and neurological exams were performed. From these exams we may conclude based on genetic and clinical features that this represents a case of EEC. These results suggest that the diagnoses of this type of clinical-pathologic entities must be done together with the involved specialists in order to establish an adequate treatment plan for the patient. Finally with detail the diagnoses and the dental treatment plan established on the child is described.
KEY WORDS: Syndrome, Dysplasia, Fissure.
Recibido para arbitraje: 24/07/2001 Aceptado para publicación: 16/1120/01
INTRODUCCIÓN
No es sino a partir de 1970 cuando Rüdiger y asociados1 aprecian que algunos de los pacientes con ectrodactilia y labio hendido tenían de manifiesto dicha asociación como un patrón sindromático en conjunción con displasia ectodérmica, designándole a este desorden el nombre de "Síndrome EEC." (Ectrodactilia-Displasia Ectodérmica-Hendidura.Esta asociación muchas veces es confundida con otro tipo de identidades quedando así erróneamente diagnosticado el paciente, por lo tanto se deben plantear los diagnosticos diferenciales del mismo. El siguiente trabajo presenta un caso clínico de un paciente masculino de 5 años de edad.
REVISIÓN DE LA LITERATURA
Cocayne en 1936 describió dos generaciones familiares con labio y paladar hendido, ectrodactilia de manos y pies y dacriocistitis, planteo que debería ser más que una coincidencia que todos los miembros de una familia tuvieran deformidad tipo pinza de langosta en las manos y labio-paladar hendido, existiendo 3 de 5 individuos afectados de esta manera. Posteriormente Clodios describe 3 casos con hendidura uní ó bilateral completa del paladar primario ó secundario, asociado con manos y pies en pinza de langosta con mal función del sistema lagrimal 3 Mc Kusick (1969) observó a una madre y a su hijo con ectrodactilia, anodoncia y obstrucción parcial del ducto lagrimal, anormalidades similares fueron reportadas por Levy (1967), lo cual designaba que la relación etiológica de estos defectos existía pero no pudieron ser precisadas para este momento1 Rüdiger y asociados en 1970 sugieren que la tríada de ectrodactilia-displasia ectodérmica-hendidura labio-palatina debería tratarse de un síndrome raro con defectos congénitos, sugiriendo el nombre abreviado de EEC.1 Bixler y asociados en un recuento de la literatura, encontraron 5 probables ejemplos del Síndrome EEC, dos de los cuales ocurrieron en familias afectadas, lo que designaría su carácter autosómico dominante, y tres de los cuales fueron esporádicos2 pudiendo representar estos últimos neomutaciones. Pries (1974) reporta un posible patrón hereditario autosómico recesivo.
CRITERIOS MÍNIMOS DE DIAGNÓSTICO
Las anormalidades son facultativas y no obligatorias por lo que el fenotipo puede variar de un paciente a otro. La ectrodactilia es la más frecuente manifestación clínica seguida de obstrucción del ducto lagrimal y labio-paladar hendido en este orden, a lo cual se suma cualquier grado de displasia cutánea-ungeal-dental y/o de cabello. La hendidura puede ser solo labial y la displasia puede ser solo discromía de cabello, cejas o pestañas.
HALLAZGOS CLÍNICOS
Los autores incluyen ectrodactilia de manos y pies; obstrucción de ductos lagrimales; fotofobia; hendidura labio-palatina; cabello ralo y escaso, cejas y pestañas con carencia parcial y/o total. Otros autores incluyen cierto grado de sindactilia de tejido blando, alteraciones albínicas de la piel y cabello; uñas hipoplásicas; ausencia de glándulas sebaceas pudiendo inexistir sudoración; numerosos nevus pigmentados; microcefalia; retardo mental; sordera; hernia inguinal; ausencia de un riñón; hidroureter; criptorquidismo; anodoncia; oligodoncia; hipoplasia del esmalte; microdoncia; caries múltiples; disminución del número de orificios Meibomianos; telecantus primario.
La piel en estos pacientes es delgada, blanca, con un grado moderado de hiperqueratosis. Las tetillas son hipoplásicas y el vello corporal puede ser escaso.
Referente a la cara, esqueletalmente es fácil visualizar una hipoplasia maxilar y en la porción media de ambos malares.16, 17, 19
COMPLICACIONES
DERIVADAS:
La ausencia u obstrucción del ducto lagrimal está asociada con lagrimeo constante, blefaritis, dacriocistitis, queratoconjuntivitis y fotofobia.
Además hay el dimorfismo buco-dental y los trastornos del lenguaje que suelen derivarse de los diferentes tipos de hendiduras3
ASOCIADAS:
Xerostomía, lengua fisurada y predisposición a sufrir de candidiasis y caries.
En raros casos se asocia retardo mental, sordera3 microcefalia, malformaciones renales e hipohidrosis, insuficiencia de la pituitaria-hipotálamo18 y a defectos del septum ventricular14
ETIOLOGÍA
El síndrome se hereda de forma autosómico dominante con penetración incompleta y expresividad variable, inclusive hay casos de etiología heterogénea o probable autosómica recesiva. La patogénesis de este defecto aún no está completamente clara, pero se cree que se debe a una translocación entre los cromosomas 7 y 9, siendo los puntos críticos a nivel del 7q11.2-q21.3 según reporte de Qumsiyeh (1992), la cual se heredaría en forma dominante y/o explicaría el alto porcentaje de casos esporádicos en las familias, supuestamente designadas hasta ahora como "neomutaciones".16 Según Celli J y cols (1999) encontraron en sus pacientes con síndrome de EEC una mutación de carácter dominante en el cromosoma P63 y en su homologo P53.
DATOS EPIDEMIOLÓGICOS
Proporción según sexo: M1:F1
Riesgo de ocurrencia: se desconoce en forma precisa pero es considerado muy bajo.
Edad de detección: al momento del nacimiento por el examen físico.
Prevalencia: rara.
Frecuencia: rara16
TRATAMIENTO
Prevención Primaria: Asesoramiento genético.
Prevención Secundaria: Estimulación del crecimiento maxilar por medio de placas ortopédicas funcionales junto con la cirugía del labio y paladar hendido. Restablecimiento del drenaje del ducto nasolagrimal y cirugías plásticas de manos y pies.
Otros Tratamientos: Tratamiento odontológico integral, ortodontico y protésico, quirúrgico, y en algunos casos terapias de lenguaje.
Momentos quirúrgicos mas adecuados para el Labio y Paladar Hendido:
I) Intervención del labio a los 3 o 6 meses.
II) Intervención del Paladar Blando a los 3 o 6 meses.
III) Intervención del Paladar Duro entre los 5 a 8 años de edad 15
Pronóstico: Bueno para la vida, en la mayoría de los casos existe un nivel de inteligencia normal; las funciones varían según la severidad de los defectos.
CONSIDERACIONES ESPECIALES
DIAGNÓSTICOS DIFERENCIALES
El síndrome EEC debe ser cuidadosamente distinguido de otras entidades. Es un síndrome autosómico dominante con penetración incompleta y expresividad variable. La hendidura de labio y ausencia del conducto lagrimal no es una combinación usual en otras condiciones.
Síndrome Rapp-Hodkin: consiste en hendidura labio-palatina, hipohidrosis, cabello delgado y alambrino, y uñas distróficas. La forma de herencia es autosómica dominante pero no hay ectrodactilia.
Síndrome Freire-Maia (Displasia Ectodérmica Tetramelica): es autosómico recesivo, la tetraperomelia está asociada con orejas largas y deformadas, pelo ralo, tetillas hipoplásicas, oligodoncia, forma de la corona de los dientes cónica, hipogonadismo y deficiencia mental.
Síndrome de Roberts: herencia autosómica recesiva. La hendidura labio-palatina y la tetrafocomelia están asociadas con hipertelorismo y agrandamiento del pene o clítoris.
Síndrome de Pseudotalidomida: la tetrafocomelia y la hendidura labio-palatina están asociadas con cartílagos hipoplásicos del ala nasal y orejas, hemangiomas faciales, y deficiencia mental.
Síndrome Autosómico Dominante De Fontaine: consiste en ectrodactilia y sindactilia de los pies, paladar hendido (usualmente submucoso), micrognacia, pabellón auditivo displásico y en algunos casos retardo mental.
Casos aislados de Ectrodactilia: pueden ser heredados de forma autosómico dominante, y menos comúnmente de manera autosómico recesivo.
Síndrome de Goltz-Gorlin: incluye además retraso en el crecimiento y neurosicomotor, microftalmia y anormalidades típicas de piel resultantes de la hipoplasia dérmica. La forma de herencia es autosómico dominante ligado al cromosoma X, por lo cual resulta letal en hombres hemicigotos en la posteridad. No hay ectrodactilia.10
Síndrome Adult (Acro-Dermato-Ungual-Lacrimal-Tooth): cursa con hipodoncia, pérdida prematura de dientes permanentes, ectrodactilia, obstrucción del ducto lagrimal, onicodisplasia y excesivas lesiones pigmentadas. Actualmente se desconoce la asociación de esta entidad con el síndrome EEC típico o si se trata de una forma del mismo síndrome.13
REPORTE DE UN CASO CLÍNICO
Pre-escolar masculino de 5 años de edad, producto de una segunda gestación de parto normal y a término, de madre normal. Se ignoran datos familiares del paciente por ser dado en adopción. Fue recibido con el diagnóstico errado de Displasia condroectodérmica.
Según reporte de la historia clínica el paciente padeció de Dacriosistitis biocular a los 7 meses por lo cual se le realizaron dos sondajes lagrimales, y posteriormente se le indicó el uso de lagrimas artificiales. Fue intervenido de hendidura labial bilateral a los 3 años y 6 meses, y a pesar de la edad actual del paciente no ha podido ser intervenido de paladar duro y blando debido a la multiplicidad de caries y focos infecciosos bucales presentes.
Debido a trastornos de conducta como inquietud y agresividad el psiquiatra lo medica con Tegretol una vez al día.
Los hallazgos clínicos muestran signos neuroectodérmicos típicos como dermatitis exfoliativa debida a la hiperqueratosis, piel delgada y blanquecina. El pelo es de color claro, escaso y delgado tanto en el cuero cabelludo como en pestañas y cejas. Al examen facial se evidencia hendidura labio-palatina bilateral completa e hipoplasia malar. (Fig.1). Intrabucalmente se corrobora el tipo de hendidura, microdoncias, oligodoncia solo de dentición permanente, con ausencia congénita de los gérmenes dentarios de ambos incisivos laterales superiores, incisivo central inferior derecho y segundos premolares inferiores. En la dentición temporal presente se evidencian caries rampantes e hipoplasias del esmalte generalizadas, a lo que se suma un grado de xerostomía leve (Fig.2).

Figura 1.: Notese la piel blanca y delgada; cabello, cejas y pestañas escasas. Achatamiento del tercio medio facial por la hipoplasia de ambos huesos malares y al tipo de hendidura.
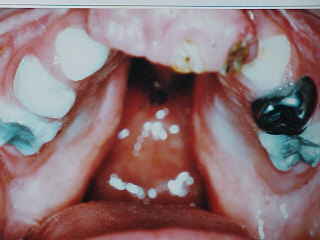
Figura 2. Hendidura labio-palatina bilateral completa, microdoncia, oligodoncia, caries rampante, hipoplasia del esmalte.
Los ojos presentan iris azul, blefarofimosis, fotofobia , defectos del sistema excretor lagrimal y la dacriosistitis biocular ya corregida.
A nivel de las extremidades se observan ectrodactilias y sindactilias de manos y pies; polidactilia post-axial en ambas manos ya intervenidas (Fig.3 y 4).
El paciente presenta leve retardo en el aprendizaje y problemas en el habla, con vocabulario escaso, propios y consecuentes de la hendidura completa ya que no ha sido intervenido el paladar secundario.
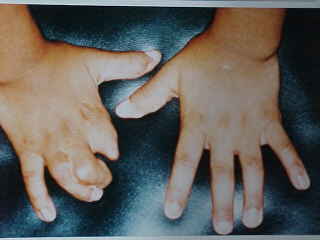
FIGURA 3: Manos con Sindactilias y ectrodactilia.
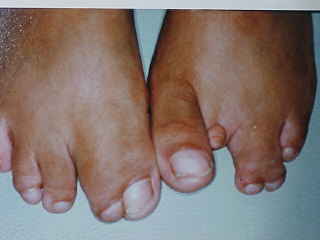
FIGURA 4: Ambos Pies con Sindactìlia y ectrodactilia.
TRATAMIENTO
El consejo genético concluye un Síndrome EEC de carácter autosómico dominante con penetrancia incompleta y expresividad variable. Se sugiere estudio urológico y oftalmológico así como protección ocular y cutánea al sol.
Con anterioridad se realizaron los drenajes de ambos ductos naso lagrímales y las cirugías plásticas en manos y pies para mejorar la función motora de los miembros y la apariencia estética de los mismos, aunque tendrá que ser sometido a otras cirugías adicionales al culminar su proceso de crecimiento y desarrollo.
Hasta el momento ha venido siendo tratado con placas ortopédicas funcionales que guían y corrigen las deformidades esquelétales presentes, facilitan la alimentación, ayuda a establecer la postura normal de la lengua y brindan ayuda psicológica a la madre adoptiva.
El tratamiento dental fue realizado bajo anestesia general debido a sus graves problemas de conducta y en sí a su síndrome, donde se le practicó en una sola sesión todo el tratamiento bucal requerido, consistiendo en múltiples pulpotomías formocresoladas, pulpectomías obturadas con pasta Maisto, banqueos con amalgamas, restauraciones cavitarias con amalgamas y resinas, y coronas de acero inoxidables cementadas con fosfato de zinc (Fig. 5).
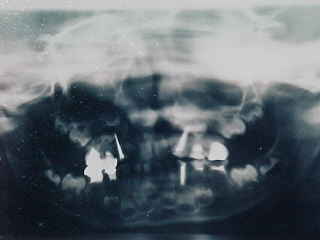
FIGURA 5: Radiografía Panorámica postoperatoria que da un aspecto global del tratamiento dental realizado durante el acto anestésico.
PRONÓSTICO
Bueno. El paciente quedó completamente restablecido, para poder ser intervenido del paladar. Se indicaron controles periódicos para terapias profilácticas y preventivas como aplicaciones tópicas de flúor, ortodoncia interceptiva y prótesis con el fin de restablecer la salud bucal, función y estética. Conjuntamente el paciente fue tratado por un terapista de lenguaje de manera de mejorar la pronunciación de las palabras y aumentar el vocabulario existente.
CONCLUSIÓN
Los pacientes con anormalidades tipo ectrodactilia, obstrucción del ducto lagrimal, labio-paladar hendido y algùn grado de displasia dental o de cabello debe ser diagnosticado con Síndrome EEC; el cual se hereda de forma autosomico dominante y debe ser corroborado por un estudio genetico.
Todo paciente con patrones anormales sistémicos requieren de una evaluación cuidadosa que involucre historia clinica, evolución del paciente y exámenes complementarios, permitiendo asì hacer hallazgos esenciales y poder diagnosticar con exactitud el tipo de síndrome que se presenta.
Este tipo de casos debe ser evaluado por un equipo multidisciplinario de genetista, odontopediatra, pediatra, oftalmólogo, nefrólogo y cirujano plástico, lo cual permite establecer un plan de tratamiento lo más acorde con los padecimientos y requerimientos del paciente.
REFERENCIAS BIBLIOGRAFICAS
1.Rüdiger,R., Haase, W., and Passarge,E. (1970). Association of ectrodactyly, ectodermal dysplasia, and cleft lip-palate. Am.J.Dis.Child., 120:160. [ Links ]
2.Bixler, D., Spivack, J., and Christian, J. (1971). The ectrodactyly-ectodermal dysplasia-clefting (EEC) syndrome: Report of 2 cases and review of the literature. Clin. Genet., 3:43. [ Links ]
3.Robinson,G., Wildervanck, L., and Chiang, T. (1973). Ectrodactyly, ectodermal displasia, and cleft lip-palate syndrome. J. Pediatr., 82:107. [ Links ]
4.Kaiser-Kupfer, M. (1973). Ectrodactyly, ectodermal dysplasia and cleft syndrome. Am. J. Ophthalmol., 76:992. [ Links ]
5.Penchaszadeh, V. And De Negrotti, T. (1976). Ectrodactyly- ectodermal dysplasia-clefting (EEC) syndrome: dominant inheritance and variable expression. J. Med. Genet., 13: 281-284. [ Links ]
6.Smith, D. (1976). Recognizable patterns of human malformation. Second edition. [ Links ]
7.Cohen, M.M. Jr. (1978). Syndromes with cleft lip and cleft palate. Cleft Palate-J. Oct., 15 (4): 306-328. [ Links ]
8.Bergsma, D. (1979). Birth defects compendium. Second edition. [ Links ]
9.Rodini ESO, Richieri-Costa A. (1990). EEC syndrome: Report on 20 new patients. Clinical and genetic considerations. Am. J. Med. Genet., 37:42-53. [ Links ]
10.Rodini, A., Nardi, M., Guión Almeida, A. And Richieri-Costa. (1992). Ectodermal dysplasia, ectrodactyly, clefting, anophthalmia/microphthalmia, and genitourinary anormalies: Nosology of Goltz-Gorlin syndrome versus EEC syndrome. Am. Med. Genet., 42:276-280. [ Links ]
11.Tanboga, Y., Pince, S. and Duzdar, L. (1992). Dental management of a child with EEC syndrome. Int. J. Pediatr. Dent., 2(2): 99-103. [ Links ]
12.Qumsiyeh, Mb. (1992). EEC syndrome (ectrodactyly, ectodermal dysplasia and cleft lip/palate) is on 7p11.2-q21.3. (Letter; comment). Clin. Genet., 42 (2): 101. [ Links ]
13.Propping, P. And Zerres, K. (1993). ADULT-Syndrome: An autosomal-dominant disorder with pigment anomalies, ectrodactyly, nail dysplasia, and hipodontia. Am. J. Med. Genet., 45: 642-648. [ Links ]
14.Ram SP; Noor AR; Ariffin NA. (1994). A neonate with ectodermal dysplasia ectrodactyly clefting syndrome and ventricular septal defect. Singapore Med J 35(2):205-7. [ Links ]
15.Kimura Takao, Atlas de Cirugía Ortognatica Maxilofacial Pediatrica, Edit. Médico-Odontológica, p35-142, (1995). [ Links ]
16.Buss PW; Hughes HE; Clarke A. (1995). Twenty-four cases of the EEC syndrome: clinical presentation and management. Med Genet; 32(9):716-23. [ Links ]
17.Moerman P; Fryns JP. (1996). Ectodermal dysplasia, Rapp-Hodgkin type in a mother and severe ectrodactyly-estodermal dysplasia-clefting syndrome (EEC) in her child. Am J Med Genet; 14;63(3):479-81. [ Links ]
18.Gershoni-Baruch R; Goldscher D; Hochberg Z. (1997). Ectrodactyly-ectodermal dysplasia-clefting syndrome and hypothalamo-pituitary insufficiency. Am J Med Genet; 68(2):168-72. [ Links ]
19.O^Quinn JR; Hennekam RC; Jorde LB; Bamshad M. (1998). Syndromic ectrodactyly with severe limb, ectodermal, urogenital, and palatal defects maps to chromosome 19. Am J Hum Genet; 62(1):130-5. [ Links ]
20.Celli J and Cols. (1999). Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99(2):143-53. [ Links ]
