










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Obstetricia y Ginecología de Venezuela
versión impresa ISSN 0048-7732
Rev Obstet Ginecol Venez v.68 n.4 Caracas dic. 2008
Insuficiencia ovárica prematura
Dra. María Scucces
Dirección: Urb. San Isidro 4ª Calle Qta. "María Teresa". Maracay e-mail:mscucce@yahoo.com Teléfono: 0243-2332256 – 0414-4581557
INTRODUCCIÓN
La insuficiencia ovárica prematura (IOP) se identifica por primera vez en 1930, pero sólo en 1950 se definen sus características clínicas. Comprende aquellas condiciones caracterizadas por la precoz desaparición de la funcionalidad ovárica, y se define IOP como amenorrea primaria o secundaria con hipoestrogenismo y elevados niveles de gonadotropinas antes de los 40 años de edad. El único criterio diagnóstico de este síndrome está representado por niveles de FSH mayores de 40 UI/L obtenidos dos veces a intervalo de un mes en una mujer de edad menor a los 40 años (1-3).
La causa de esta enfermedad permanece oscura en la mayoría de las pacientes. Es un desorden heterogéneo cuya etiología incluye el amplio espectro de las disgenesias gonadales y otras causas de origen no disgenético. En todas ellas, las sintomatología clínica es muy parecida a la observada en la menopausia fisiológica: amenorrea, elevados niveles plasmáticos de FSH (hipergonadotropismo) e hipoestrogenismo (1,2,4-5).
La incidencia de IOP es altamente variable de acuerdo al contexto familiar y a la sintomatología clínica que conduce al diagnóstico y ha sido reportado que puede variar entre el 0,9 % y el 3 % de la población general (4,6-8). La prevalencia de IOP en todos los casos de amenorrea, varía entre 4,8 % y 10 % en función del límite de edad al cual se atribuya (30, 35 ó 40 años). Otros autores reportan una prevalencia entre el 10 % y 28 % en pacientes con amenorrea primaria y del 5% al 10 % en pacientes con amenorrea secundaria. Además se reporta una incidencia del 3 % en la población femenina, del 1 % en las mujeres de 40 años y 0,1 % de IOP en 2 373 pacientes posmenopáusicas estudiadas (10). Si bien estos límites de edad han sido establecidos arbitrariamente, están fuera de las dos desviaciones estándar de la edad media de la menopausia fisiológica que se ubica alrededor de los 51 años (1,2,9-11).
El compromiso del factor ovárico después de la menarquía, pero antes de los 40 años de edad se conoce también como "menopausia precoz". La mayor parte de los casos de IOP no se identifican sino por su baja fertilidad. La escasa influencia de los factores reproductivos y de los hábitos de vida sugiere que el factor genético juega un rol importante y ello trae como consecuencia que el tratamiento se limite esencialmente a instaurar una terapia hormonal de reemplazo, que tienda a evitar las complicaciones a mediano y largo plazo de la carencia estrogénica y a evaluar, caso por caso, una eventual procreación. Cuando la enfermedad se presenta en pacientes jóvenes, ellas poseen un 5 % - 10 % de la probabilidad de obtener embarazos espontáneos; por tanto, el cese de la actividad ovárica puede no ser irreversible aun cuando los niveles de FSH estén transitoriamente elevados y una residual función folicular puede que aún esté presente (2,12-21). En el Cuadro 1 se reporta la clasificación de la IOP (2).

ASPECTOS EMBRIOLÓGICOS
Las gónadas poseen un origen común con el riñón primitivo (mesonefros) (22). Tres semanas después de la fecundación, las gónadas aparecen en la pared posterior de la cavidad celómica e independientemente del sexo genético, se originan de dos derivados del mesodermo (epitelio celómico y mesénquima) las células germinativas primordiales. En la 4ª semana, sobre la cara anterior del mesonefros, y a cada lado de la línea media, se desarrollan los pliegues o crestas genitales o gonadales, por un engrosamiento del epitelio celómico y por condensación del mesénquima subyacente. En la 5ª semana las células germinativas migran del endodermo, siguiendo el mesenterio dorsal del intestino posterior, a la cresta genital. Estas células se denominan oogonias. Para la 6ª semana las células germinativas primordiales, formadas en la pared del saco vitelino cerca del alantoides, son transportadas por el desplazamiento de las hojas embrionarias y mediante movimientos ameboideos, completan su migración en las crestas genitales. Se forma así la gónada indiferenciada. En ausencia de tal penetración, ningún ulterior desarrollo de la misma puede tener lugar, siendo necesaria la presencia de dos cromosomas X intactos para que las oogonias puedan entrar en meiosis y formar los oocitos primarios (22,23).
Entre la 7ª y 8ª semana, en el embrión con genotipo 46,XX, el futuro ovario puede identificarse sólo por ausencia de las características morfológicas del testículo. A la 8ª semana el número de oogonias es de 600 000 células. A finales de la misma, se inician y se verifican en la gónada, hasta esa época "indiferente", mutaciones características: a) Los cordones sexuales primitivos son disgregados por proliferaciones mesenquimales, en acúmulos regulares aislados. b) Involución rápida e incompleta de las conexiones existentes entre los cordones primarios y los tubos del mesonefros, y de las que permanecen algunos vestigios: epoóforo paroóforo, rete ovaris y células hiliares.
Entre la 8ª y 11ª semana, la gónada aumenta de volumen y asume una configuración fusiforme a lo largo de la pared abdominal interna. Al final del primer trimestre de la gestación, la presencia de un cariotipo apropiado condiciona la diferenciación sexual de la gónada. Hacia la 11ª semana, las oogonias que están ubicadas en la proximidad del mesonefros, cesan su actividad mitótica, mientras que las que están en la porción cortical continúan su actividad por algunas semanas (2,22,24,25).
En el período fetal las oogonias carecen de cobertura celular. A las 20 semanas de gestación las oogonias de la región medular interna del ovario detienen su meiosis, iniciada ya a la 12ª semana de gestación, en la etapa de diploteno de la profase de la primera división meiótica. Las oogonias de la meiosis se denominan ahora oocitos, y están rodeados por células foliculares de la granulosa de forma aplanada que descansan sobre una membrana basal. Se forma así el folículo primordial y permanecerá así en el resto de la vida fetal, en el recién nacido y en la vida prepuberal hasta que no sea reclutado en un nuevo ciclo en la pubertad (22).
De los 7 millones de oogonias en los ovarios de fetos femeninos a las 20 semanas de gestación, al nacer sólo existen 2 millones de oocitos. La mayoría de los folículos humanos están destinados a la atresia, fenómeno que se relaciona con la muerte celular programada o apoptosis, de allí que en la pubertad sólo queden 400 000 oocitos, y de ellos sólo 500 oocitos o menos serán ovulados. La cantidad total de folículos primordiales disminuye por debajo de 100 000 en la edad reproductiva. A partir de la 4ª década de la vida la depleción folicular se acelerará y se agotará con el advenimiento de la menopausia (12,22,26).
FACTORES ETIOLÓGICOS
Insuficiencia ovárica prematura por menor dotación folicular
Deficiencia inicial del número de folículos
En estudios realizados sobre animales, varios genes han sido implicados en la patogénesis de la IOP. El gen c-kit en el locus white spotting (W) codifica un receptor de la tirosinasa que se liga al factor KL, codificado, a su vez, por el locus SI (stem cells). Mutaciones de uno u otro locus determinan la no migración de diferentes líneas celulares y, entre ellas, de las células germinales, con el consiguiente cuadro de insuficiencia ovárica, alteraciones de la pigmentación y anemia (4), además el síndrome germ cell deficient (gcd), de carácter autosómico recesivo, ocupa el locus situado en el cromosoma 11/A2-3 y se caracteriza por la falta de migración y/o proliferación de las células germinativas (1,2,27). La timectomía en el mono Rhesus se asocia a una disminución del número de oogonias, no alterando su proceso de maduración (2).
En humanos, el gen ATM (ataxiatelangiectasia) codifica proteínas implicadas en el mantenimiento del "pool" de folículos primordiales (27).
Atresia folicular acelerada
Este proceso se observa en algunas anomalías cromosómicas. La muerte celular programada tiene un rol fundamental en el proceso de atresia folicular. Existen factores que la inhiben y otros que la estimulan y, ambos, actúan sobre la expresión del gen BCL-2 que parecería ser una de las claves reguladoras de la muerte celular programada (1,2).
Cromosoma X
El cromosoma X juega un rol importante en el proceso de dotación folicular y en el control de la función ovárica. Su parte distal posee numerosos genes que podrían estar implicados en la función reproductiva (2).
El síndrome del cromosoma X frágil está situado en el brazo largo (Xq) en el locus 26-27 (Xq 26-27). En el locus FRAXA, el gen FMR1 codifica tripletas CGG. La premutación de la zona conocida como FRAXA frágil determina la repetición de las tripletas CGG en frecuencias que pueden alcanzar hasta las 200 veces. Las pacientes portadoras de esta premutación desarrollan en el 16 % de los casos IOP, rasgo éste que las diferencia de las portadoras de la mutación completa pues en ellas no se manifiesta IOP. Si bien se desconoce el mecanismo de asociación entre IOP y la premutación FRAXA, ésta tiene valor como marcador genético en la identificación de familias a riesgo para la transmisión del síndrome del cromosoma X frágil. Las pacientes portadoras de la premutación FRAXA poseen niveles de FSH significativamente elevados lo cual es un indicador de riesgo de IOP y se traduce en importantes implicaciones para la fertilidad en las mismas, en estas pacientes han sido identificadas microdelecciones del gen FMR2 localizado en el locus FRAXE (2). Es importante el estudio familiar en aquellas portadoras de la premutación FRAXA con la finalidad de prevenir la transmisión del síndrome (2,28-34).
La polisomía X (47XXX) y el cromosoma X en anillo se pueden asociar a IOP (29). Asimismo, se ha podido evidenciar la presencia de translocaciones balanceadas entre un cromosoma autosómico (autosomas 6, 16, 20 y 22) y el cromosoma X. No se comprende bien por qué aparece IOP en la translocación y se formulan diversas hipótesis.
En cambio, han sido evidenciadas zonas críticas del cromosoma X a nivel de su brazo largo Xq13.3–q21.3 y de Xq 26-28 y, más aún, se ha identificado el gen POF-1 localizado en Xq21.3-27 y el gen POF-2, ambos de origen paterno, implicados en la IOP (1,2,14,29,35-37). Un grupo italiano ha descrito recientemente un análogo del gen "dia" en el hombre. Dicho gen, al mutar, produce esterilidad tanto en el macho como en la hembra de la Drosophila. En el hombre, el gen se ubica en la parte distal del Xq21 que codifica una proteína de 361 aminoácidos (proteína dia). Mutaciones en los alelos del gen afectan la espermatogénesis o la ovogénesis conduciendo a la esterilidad. El gen (dia) se expresa en las primeras fases del desarrollo embrionario cuando se forma la gónada, y la proteína que codifica posee dos dominios FH1/FH2 (formin homology 1 y 2). La proteína "dia" pertenece a una familia más amplia de proteínas implicadas en la citoquinesis y en la organización del citoesqueleto. En una paciente con cuadro clínico de amenorrea secundaria a los 17 años y con desarrollo puberal normal se ha evidenciado una translocación 46,Xt (X;12) )q21, p13) con ruptura del último intrón del gen dia (2,38).
Otras de las anomalías estructurales están representadas por la delección, total o parcial, del brazo corto del cromosoma X (Xp-) o de un brazo largo (Xq-) del mismo. Los aspectos clínicos de estas pacientes son similares a los del síndrome de Turner pudiendo presentar enanismo, gónadas rudimentarias (streak gonad) y otros síntomas de la disgenesia gonadal, además pueden presentar tiroiditis de Hashimoto y diabetes. En el 25 % de estos sujetos la anovulación y la amenorrea se verifican después de un período variable de ciclos menstruales más o menos regulares (2,24,42,48).
Marozzi y col. (39), describen 6 casos de pacientes con IOP portadoras de una delección del brazo largo del cromosoma X (Xq). En todas ellas estaban ausentes los estigmas morfológicos turnerianos. La delección de una restringida zona del brazo largo del cromosoma X (Xq) puede ser responsable del fenotipo para IOP, y dicha región podría estar ubicada entre Xq26.2 a Xq28 abarcando aproximadamente 22 Mb de ADN. Otro caso, reportado por Davison y col. (40), con delección Xq 26.1 resultó con historia de familiaridad para la delección parcial del cromosoma X, su diagnóstico precoz impidió la instauración IOP así como permitió la implementación de estrategias para la fertilidad (2,39-43).
Disgenesia gonadal pura
La asociación entre un cariotipo normal (bien sea 46XX o 46 XY) y gónadas disgenéticas, definen la disgenesia gonadal pura (24). Los sujetos pertenecientes al grupo de las disgenesias gonadales puras, son fenotípicamente normales, a excepción de la ausencia de caracteres sexuales secundarios que le imprimen un aspecto infantil (48).
Los sujetos con disgenesia gonadal XY (síndrome de Swyer) son fenotípicamente mujeres con falta de desarrollo de caracteres sexuales secundarios, y presencia de estrías gonadales que histológicamente recuerdan vestigios testiculares funcionantes (24).
La relación entre la presencia del cromosoma Y y la diferenciación testicular aún permanece oscura. Han sido propuestos varios factores determinantes testiculares (TDF), entre los que está el gen Sry localizado en el brazo corto del cromosoma Y (22). El primero de los TDF propuestos, en cambio, fue el antígeno H-Y, ubicado en el brazo largo del cromosoma Y. Los casos con positividad serológica para este antígeno poseen elevado riesgo para el desarrollo de gonadoblastomas (22,42,44).
En el ámbito de las disgenesias gonadales 46 XY, la evidencia de estructuras müllerianas y los niveles de testosterona dentro de límites normales para la mujer, diferencian al síndrome de Swyer, del cuadro del síndrome de insensibilidad congénita a las andrógenos (síndrome de Morris)
el cual es debido a una insensibilidad periférica a los andrógenos y se corresponde con la ausencia de receptores para los mismos. Se hereda en forma autosómica recesiva ligado al X. Su diagnóstico debe inducir a investigar la detección de otros casos en el grupo familiar, en efecto, en 2001 Caraballo Mata y col. describen el caso de tres hermanas consanguíneas portadoras del síndrome de Morris (24,42,45,46).La disgenesia gonadal pura con cariotipo 46XX se caracteriza por un fenotipo femenino, es decir, normal. Las gónadas están representadas por bandas fibrosas que a la biopsia resultan carentes de folículos (43,47,48). La ausencia de los folículos parece ser consecuencia de una mutación autosómica/somática en los genes que controlan la migración de las células germinales o a la proliferación mitótica de las oogonias
(1).Disgenesias gonadales con anomalías de los cromosomas sexuales
El síndrome de Turner representa el prototipo de este grupo de disgenesias gonadales. El espectro de alteraciones genéticas comprende esencialmente los siguientes tipos: 45,X0; 46X,i(Xq); 45X0/46XX; 45X0/46XY; 45X/46,i(Xq), la insuficiencia ovárica en este grupo de pacientes sería consecuencia de una degeneración de folículos primordiales que, precozmente, finalizan la primera división meiótica y este efecto se debe a la ausencia del locus en los brazos largo y corto del cromosoma X, lo cual sería responsable de un inadecuado revestimiento de las células de la granulosa con la consiguiente producción inadecuada de factores inhibidores de la meiosis
(41,42). En el 75 % de las pacientes con monosomía X, el cromosoma sexual proviene de la madre y la anomalía del cariotipo es de origen paterno por la fecundación de un oocito normal por parte de un espermatozoide con nulisomía X o Y. El origen es también paterno, en aquellos casos de cromosomas anormales por delección del brazo largo del cromosoma X y la ausencia de material genético en el area pericéntrica de los brazos largos del mismo, representa la condición que determina la anomalía estructural ovárica (24,49). El fenotipo de los sujetos con síndrome de Turner es femenino y su incidencia es de 1 por cada 25 000 a 27 000 recién nacidas vivas. La anomalía somática más constante es la baja estatura que se mantiene por debajo del 3º percentil, además, otras manifestaciones clínicas incluyen el infantilismo sexual, útero y vagina hipoplásicos, amenorrea primaria, braquicefalia, pterygium colli (rasgo indispensable para algunos para el diagnóstico del síndrome de Turner típico), cubitus valgus, micrognatia, tórax "en escudo", epicanto, ptosis palpebral, paladar hendido, implantación baja del cabello y del pabellón auricular, acortamiento del cuarto metacarpiano, linfedema de manos y pies, otitis media recurrente que puede conducir a la sordera neurosensorial, vicios de refracción, nevus pigmentados, hipertensión. Pueden estar presentes también anomalías somáticas más graves como lo son las malformaciones cardiovasculares como la coartación de la aorta y renales como el riñón en herradura, la aplasia renal (23,24,29,42,48,49). En el 2000 Terán Dávila y col. describen un caso de una paciente con síndrome de Turner puro, pero con baja expresión de los estigmas somáticos turnerianos, normal desarrollo de los caracteres sexuales secundarios, menarquía espontánea a los 13 años y ciclos menstruales regulares por diez años (49).En el ámbito de las disgenesias gonadales es posible encontrar variantes que son resultado de anomalías cromosómicas y el mosaicismo es responsable de la mayor parte de estas variantes de disgenesia gonadal.
En los sujetos con disgenesia gonadal que presentan un desarrollo gonadal asimétrico, con un testículo de un lado y una gónada rudimentaria (
streak gonad) del otro lado se habla de disgenesia gonadal mixta, ya descrita por Shoval en 1953 (50). El fenotipo más frecuente es el femenino en 66 % de los casos con una diferenciación masculina más o menos acentuadadel tracto genital
(47,48,50,51). Se desconoce la etiología de esta diferenciación sexual (50). La forma más común de mosaicismo en los pacientes con este síndrome es el tipo 45,X0/46,XX; otras variantes de mosaicismo se caracterizan por cariotipo 45,X0/47,XXX y 45,X0/46,XX/47,XXX. Un 6 % de las mujeres tiene un mosaicismo y presencia de un cromosoma Y en su patrimonio genético y manifiestan un cariotipo 45X0/46XY, que es el más frecuente o bien 45,X0/47,XYY o 45,X0/46,XY/47,XYY (48,50). Las pacientes con mosaicismo y presencia de un cromosoma Y normal o estructuralmente alterado en su patrimonio genético, tienen una incidencia de transformación neoplásica maligna de las gónadas (germinomas, gonadoblastomas) elevada que alcanza casi el 20 % al 27,5 %. El gen responsable del gonadoblastoma (GBY) ha sido identificado cerca del centrómero del cromosoma Y. La técnica de hibridización in situ fluorescente (FISH) con prueba específica centromérica es de utilidad para evaluar el riesgo de gonadoblastoma (48,50). La gonadectomía profiláctica es una indicación absoluta en todos aquellos casos en los que se identifique la presencia de material cromosómico Y (24,50). Los sujetos con disgenesia gonadal y mosaicismo cromosómico que comprenda al cromosoma Y (45X0/46XY), pueden presentar una ambigüedad sexual de entidad variable relacionada con la presencia de actividad testicular. La asimetría en las anormalidades genitales es debida al predominio de las líneas celulares con cariotipo 45X0 en la gónada bandeleta y 46XY en el testículo disgenético (24,42,50).Los sujetos con disgenesia gonadal asociada a un defecto de las gónadas con presencia de isocromosoma en el brazo largo de X 46,i(Xq) poseen elevada frecuencia de padecer tiroiditis autoinmune (tiroiditis de Hashimoto) y carcinoma tiroideo (42,48).
Cromosoma 3
Existe relación genética entre mutaciones en el brazo largo del cromosoma 3 (3q21-24) y el síndrome de blepharophimosis (disminución del borde palpebral, ptosis palpebral y epicanto inverso). Los genes implicados en este síndrome, codifican proteínas involucradas en la atresia folicular
(2,27,41,52).Galactosemia
Representa un síndrome muy raro, de carácter familiar, transmitido en forma autosómica recesiva. Se caracteriza por el déficit de galactosa-1-fosfatouridil–transferasa, y, la IOP se asocia a nefropatía grave. Se desconoce el porqué de la IOP en la galactosemia. Los modelos animales sugieren una reducción del número de oocitos, sin embargo, los preparados histológicos de ovarios de las pacientes afectadas, no confirman su existencia. Ha sido identificado un marcador genético (GALT 188Q) en pacientes con galactosemia, el cual está asociado a un riesgo, si bien bajo, de disfunción ovárica en aquellas mujeres heterocigotas para la mutación. Finalmente, la galactosemia representa un buen ejemplo de la dificultad para discriminar la etiología de la IOP entre formas por depleción y formas por disfunción folicular (2,27,29,43).
Quimioterapia y radioterapia
La quimioterapia y la radioterapia como causas de IOP, pueden variar en el tiempo. Los medicamentos que pueden producir IOP se indican en el Cuadro 2, en cambio, en la Figura 1 se representa un esquema que evidencia, durante la quimioterapia, cualquier evolución posible de IOP (2,43).
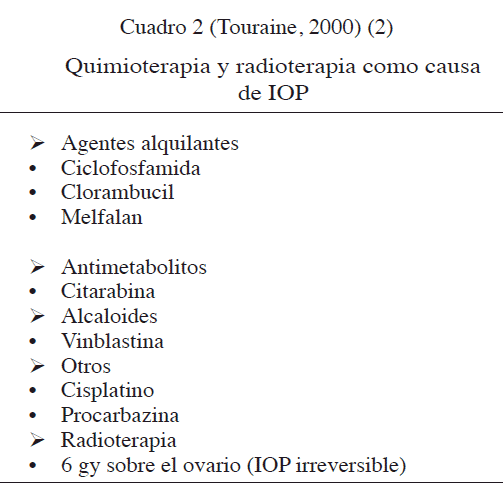
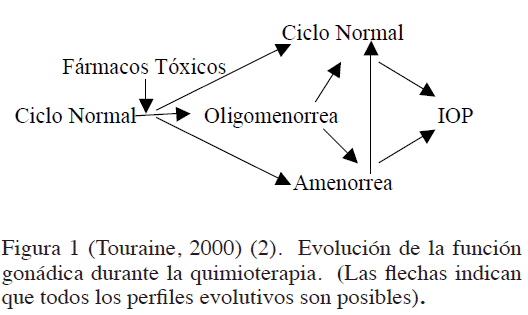
Aunque la IOP a los 30 años es más fácil-mente reversible que a los 40 años, en ningún caso es posible manifestar a la paciente certeza alguna. El período de máximo efecto de la quimioterapia, probablemente, preceda el reinicio de la meiosis. La susceptibilidad del parénquima ovárico al medicamento parece estar directamente relacionado con la edad de la mujer y con la dosis (2,24,43,53).
Por otra parte, dosis de radiaciones ionizantes de 150 rads que interesen el área pélvica, pueden afectar la función ovárica sólo en mujeres de edad superior a los 40 años. Dosis mayores a los 700 rads inducen un daño ovárico irreversible en 100 % de los casos, independientemente de la edad. Las radiaciones ionizantes y la quimioterapia pueden producir daños al ADN que resultan en muerte celular o daños subletales (2,24,43,54,55).
Entre las pacientes fumadoras ha sido posible identificar una menopausia precoz. Se señala en ellas, como causa de IOP, una mayor frecuencia de diploidía ovocitaria por incapacidad de expulsión del primer corpúsculo polar en la meiosis (1).
Iatrogénicas
Entre las causas iatrogénicas de falla ovárica han de mencionarse todas aquellas "manipulaciones" quirúrgicas como son las resecciones ováricas, la enucleación de los quistes de ovario y otras intervenciones imprudentes (2,43).
Autoinmunidad
La acción de la autoinmunidad puede determinar bien una reducción del número de folículos así como una destrucción acelerada de la funcionalidad ovárica (1). Es conocida la presencia de anticuerpos antiovario, contra los receptores de las gonadotropinas, contra los esteroides y la zona pelúcida (1).
Se puede asociar IOP a patologías autoinmunes como son la enfermedad de Addison, la tiroiditis de Hashimoto, el hipoparatiroidismo, la enfermedad de Graves, la diabetes juvenil, la miastenia, la enfermedad de Crohn, el vitiligo, el lupus eritematoso sistémico (LES) y la artritis reumatoide, el síndrome de Sjögren, la anemia hemolítica autoinmune y la púrpura trombocitopénica idiopática (1,24,43,56). Estos casos se caracterizan por una activación de los linfocitos T, que, a nivel ovárico, se manifiesta con una importante infiltración de la teca, en modo particular de aquella de los folículos maduros, lo cual sugiere, la implicación de las gonadotropinas como lo es la FSH en la reactividad antigénica (2,43).
La patogénesis autoinmune está obstaculizada por el diagnóstico, siendo los anticuerpos antiovario, poco específicos de tal forma que algunos autores cuestionan su utilidad clínica, mas sin embargo, su presencia puede jugar un rol o reflejar un proceso autoinmune ovárico responsable del desarrollo de IOP (2,9,57-60). Los anticuerpos contra la 21-hidroxilasa, en cambio, son altamente específicos en la enfermedad de Addison idiopática, y sus niveles disminuyen a medida que aumenta la duración de la enfermedad (24,61).
Recientemente ha sido descrita IOP en el cuadro de un síndrome APS-1 (síndrome poliglandular autoinmune tipo 1) ligado a mutaciones del gen APECED (Autoinmune Polyendocrinopathy-Candidiasis-Ectodermal-Dystrophy), que codifica un particular factor de transmisión cuyo rol se desconoce y no pertenece al sistema HLA. La presencia de anticuerpos contra varios órganos endocrinos causa una insuficiencia pluriglandular, que, en la mayoría de los casos, está caracterizada por hipoparatiroidismo, enfermedad de Addison e IOP (2,62,63). En el síndrome de deficiencia poliglandular autoinmune tipo II se asocian el hipotiroidismo, la deficiencia suprarrenal y la IOP (29). El trastorno reproductivo puede anteceder de varios años la aparición conclamada de la patología autoinmune pluriglandular (24).
El proceso central de la atresia folicular en la autoinmunidad lo es la activación cíclica de la cascada de las citoquinas iniciada por el interferón–gamma que induciría la producción continua, de citoquinas como mecanismo clave en la patogénesis de la IOP (9,64). Los datos que soportan la etiología autoinmune de la IOP incluyen: a) infiltración ovárica por linfocitos y células plasmáticas y alteraciones en subclases de células T, b) circulación de anticuerpos antiovario, c) recuperación de la función ovárica con la regresión del desorden autoinmune (1,2,9,65-68).
Agentes virales
Las pacientes afectadas por el virus de la parotiditis pueden presentar ooforitis en 5 % de los casos, la cual puede ser responsable de IOP. Se señalan también
las infecciones por Coxsackie y rubéola como causa de IOP (1,29,43).
Insuficiencia ovárica prematura por disfunción folicular
Factores ovocitarios
Las células de la granulosa, secretan sustancias no esteroideas de naturaleza proteica, que regulan localmente la función ovárica (acción intraovárica) o que controlan las actividades secretorias gonadotropas hipofisarias (acción extraovárica)
(2,24,69). En tal sentido, la inhibina es una glicoproteína de p.m 10.000 D, secretada por las células de la granulosa que resulta fundamental en el mantenimiento del FSH en un nivel basal. Mutaciones en el gen que la codifica pueden estar implicados en la etiología de la IOP. El factor de crecimiento y diferenciación (growth differentiation factor GDF-9) comienza a producirse en las fases precoces de la formación del folículo primario y prosigue hasta la ovulación. Desempeña un rol importante en la interacción entre oocito y folículo (2). La inactivación del gen que codifica el GDF-9, ha evidenciado una alteración en la evolución meiótica de los ovocitos, con la consiguiente formación de ovarios más pequeños, folículos primarios que no maduran y ausencia de cuerpos lúteos (2).La unión intercelular entre oocito y las células de la granulosa del tipo de los complejos de unión (gap junctions) requiere de la participación de una proteína (conexina 37) cuyo rol resulta interesante. En efecto, la inactivación del gen que codifica esta proteína, determina la desaparición de tales complejos, asociándose a una maduración folicular que no va más allá del estadio antral y una luteinización precoz con formación de cuerpos lúteos pequeños. La luteinización inadecuada de los folículos de de Graaf representa el principal mecanismo fisiopatológico de la IOP en pacientes cuyo cariotipo es normal
(1,2,27,70).Deficiencia de enzimas
Entre las causas de IOP por déficit de enzimas de la esteroidogénesis las más frecuentes son los debidas a la colesterol-desmolasa, la 17∝-hidroxilasa, la 17-20-desmolasa, y algunas aromatasas. Todos se traducen en una alteración en la regulación de la síntesis de estrógenos. Los individuos de sexo femenino que resultan afectados presentan infatilismo de las estructuras müllerianas y la ausencia de desarrollo puberal representa la condición de exordio de la enfermedad con elevados niveles de gonadotropinas, aunque haya una, aparente, normal existencia de folículos primordiales (1,24,29,43).
Defectos específicos de las gonadotropinas o de sus receptores o proteína G defectuosa
Aquellas alteraciones de la estructura de las gonadotropinas o bien de sus receptores o de la proteína "G" pueden producir IOP (1,2,29,71).
Alteraciones del gen del receptor del FSH (R-FSH) han permitido comprender mejor su rol en el proceso de maduración folicular. Corresponde a la escuela finlandesa describir por primera vez la existencia de una doble mutación homocigótica (Ala 189 Val) del gen del receptor del FSH ubicado en el hombre en el cromosoma 2. Estudios sucesivos hechos por Touraine (2)
identifican dos nuevas mutaciones de dicho gen siendo ambas pacientes heterocigóticas, la primera de ellas tiene la mutación Ile 160Thr y Arg 573 Cys; la otra paciente presenta la mutación Asp 224 Val y Leu 160 Thr (2,29).Estudios clínicos realizados en pacientes con enfermedades ginecológicas, demuestran que las que son portadoras de una variante de la LH con mutación de la subunidad beta (Trp8-Arg8 y Ile15) poseen desórdenes reproductivos, incluyendo IOP, con mayor frecuencia de las que poseen un LH normal. Las gonadotropinas hipofisarias circulan en diferentes formas moleculares, con actividad biológica diferente y la proporción de cada variedad se relaciona con la concentración de estrógenos circulantes. En los casos de insuficiencia ovárica de larga duración como ocurre en la menopausia precoz predominan formas moleculares grandes que poseen menor actividad biológica y hay una gran alteración en el perfil de la LH (29,71).
Idiopática
En la mayoría de los casos no es posible identificar la etiología de la IOP por lo que se la define como idiopática (IOP con cariotipo euploide)
(1,7,43). Falsetti y col. describen 40 casos de IOP post-puberal de la cuales el 52,5 % resultó idiopática (56). No es posible en pacientes con IOP idiopática identificar el rol de la autoinmunidad (43,74).El síndrome de Savage o síndrome del ovario resistente, ha adquirido un rol importante entre las formas de IOP idiopáticas
(1). Fue descrito por vez primera por Jones y de Moraes-Ruehsen en 1969 (29). Se caracteriza por una normalidad histológica del aparato folicular, y caracteres sexuales secundarios y cariotipo normales con elevados niveles degonadotropinas
(1,24). El defecto congénito (gen autosómico recesivo) o adquirido (autoinmune) de los receptores para la FSH de la membrana de las células de la granulosa es la hipótesis patogenética que, posiblemente, esté a la base del síndrome, y sea responsable de la refractariedad ovárica a las gonadotropinas (24,29,42).La presencia de IOP idiopática familiar puede variar de 4 % a 31 %. El estudio del pedigree de las familias afectadas muestra un patrón de herencia autosómico dominante o uno ligado al cromosoma X (X-linked) (7). En las familias con transmisión materna, bien sea que el patrón de herencia esté ligado al X o que sea autosómico, el riesgo de recurrencia de IOP es del 50 %. Por el contrario, en aquellas en las que la transmisión es paterna, el riesgo de recurrencia es del 100 % si el desorden posee un patrón de herencia ligado al X y decrece al 50 % cuando el patrón de herencia es autosómico dominante (7). De allí que, una adecuada historia familiar con análisis del pedigree, permite evaluar el riesgo que poseen las mujeres de desarrollar IOP, pudiendo éste variar desde 100 % (en los casos de IOP familiar) o ser tan bajo como 1% en los casos esporádicos de IOP. Ante la presencia de una historia familiar de IOP se recomienda el consejo genético (2,7,72-74).
ESTRATEGIAS DIAGNÓSTICAS
El diagnóstico de IOP resulta fácil si se hace una cuantificación de los valores de la FSH para evaluar la amenorrea. El criterio más utilizado para diagnosticar la IOP es la presencia de amenorrea de más de 4 meses de duración y su asociación con niveles plasmáticos de FSH de 40 pg/mL obtenidos dos veces a distancia de un mes en una mujer con edad inferior a los 40 años
(1,2,29,42,43,64). Una atenta historia clínica con una anamnesis detallada y un minucioso examen físico pueden orientar acerca de patologías específicas o no, asociadas a IOP (1).El análisis del cariotipo se impone para demostrar una anomalía cromosómica o también una translocación. La identificación de un cromosoma Y, o de fragmentos del mismo sugieren la investigación citogenética en otros sujetos de la familia que puedan estar potencialmente afectados. En aquellas pacientes cuya edad es inferior a los 35 años está indicado el análisis del cariotipo, que podrá extenderse a otros miembros de la familia (1,3,29).
En las pacientes con cariotipo normal e IOP espontánea que deseen fertilidad está justificado indagar sobre hipotiroidismo y diabetes mellitus
(1). Otras endocrinopatías han de investigarse sólo si existen datos sugestivos (insuficiencia suprarrenal, hiperparatiroidismo, anemia perniciosa, lupus y artritis reumatoide) (1,16,19,29,71-76).La evaluación inmunológica con la identificación de anticuerpos antitiroides, antinúcleo y antiovario representan un razonable abordaje diagnóstico de la anovulación hipergonadotrópica, pero en el caso de los anticuerpos antiovario, la escasa especificidad y/o sensibilidad de las técnicas hace que su determinación sea poco útil para el diagnóstico etiológico (4,16,67,77).
La ecografía pélvica y la biopsia ovárica no dan indicaciones pronósticas, pues la presencia o no de folículos no indican fertilidad en la paciente, aunque evalúen la actividad folicular ovárica. La biopsia ovárica raramente representa la condición anatómica real de toda la gónada y es un procedimiento que no está exento de riesgos y su utilidad se limita en el plano diagnóstico sólo si se programan otras investigaciones destinadas a precisar los genes potencialmente implicados en la génesis de la IOP. Se describen en la literatura embarazos con biopsia que han reportado ausencia de folículos en la gónada
(1,6,29,43,78-82).En pacientes con hipogonadismo hipergonadotropo que presenten síntomas neurológicos, es posible sospechar tumores hipofisarios productores de gonadotropinas, si bien son muy raros en pacientes con edad inferior a los 40 años. Una prueba de TRH y la resonancia magnética nuclear permiten confirmar su presencia (1).
TRATAMIENTO
Cualquiera que sea la causa de IOP, debe darse a la paciente una información precisa, diferenciando la terapia de reemplazo hormonal (TRH), de un posible procedimiento diagnóstico o terapéutico para la infertilidad
(2,8,29,43,83-86). A pesar de la posibilidad de preservar la actividad folicular, la probabilidad de embarazo en estas pacientes es realmente muy baja y la inducción de la ovulación resulta ineficaz. Los efectos sistémicos de la falla ovárica permanecen aún poco comprendidos (16). Una vez diagnosticada la IOP, esta condición clínica representa una clara indicación para la TRH a largo plazo aun en ausencia de los síntomas por déficit de estrógenos. Ha de realizarse en total acuerdo con la paciente (3,4,18,19,29,43,79,86).La menopausia precoz es una condición de alto riesgo para la osteoporosis y la enfermedad cardiovascular, mientras que está reducido el riesgo
de cáncer mamario en estas pacientes
(29,86-88). En general no hay datos disponibles acerca de los riesgos relacionados con el uso de la TRH en estas pacientes (88).El concepto de tratamiento en la IOP implica la restauración, preservación y sustitución de la función ovárica aun cuando no existe ninguno que detenga el proceso de involución ovárica. Pero la IOP puede ser, sin embargo, un proceso reversible o bien puede responder a modalidades terapéuticas, en pacientes seleccionadas (12,29,43).
En términos generales, en el caso de las neoplasias ginecológicas como el cáncer de vulva y vaginal, la TRH no está contraindicada
(1). En el cáncer de ovario la TRH no influencia negativamente la sobrevivencia de la paciente, al igual que en aquellas tratadas por carcinoma cervical y está indicado el uso de una TRH combinada continua (1). En las pacientes tratadas por carcinoma endometrial existe acuerdo en contraindicar la prescripción de la TRH (88).En el caso de neoplasias no ginecológicas la TRH debe basarse principalmente en los datos del laboratorio (presencia de receptores esteroideos en el tejido tumoral) o bien ha de hacerlo sobre la base del riesgo de insurgencia del tumor en aquellas pacientes que ya asumen TRH. En atención a estas consideraciones las pacientes, aparentemente curadas, por carcinoma tiroideo, melanoma, leucemia y linfoma pueden ser tratadas con TRH. La TRH reduce la incidencia de carcinoma de colon y la morbimortalidad con él relacionada así como la familiaridad para este carcinoma no la contraindica. En cambio, en las pacientes con meningioma, dada la elevada expresión de receptores para la progesterona que está presente en estos tumores, está contraindicado el uso del tratamiento de reemplazo hormonal (89-91).
Los datos disponibles sobre TRH y riesgo de cáncer de mama son confiables. La TRH de corta y media duración no modifican el riesgo oncológico sino que sólo actúan sobre patología distrófica y vasomotora. Beneficios para el metabolismo óseo y cardiovasculares se obtienen con tratamientos a largo plazo (65-70 años), siendo probable, si bien modesto, un aumento del riesgo de tumor mamario
(1), pero, en terapias prolongadas (mayores de 5 años) el riesgo de cancer mamario es inferior a la de otros factores etiológicos como lo son la edad de la menarquía, la del primer embarazo o la familiaridad (88). No puede darse indicación acerca de la conducta a seguir en cuanto a TRH en las pacientes portadoras de mutaciones de los genes BRCA (1). En la literatura, existe acuerdo general sobre el hecho de que los estrógenos solos no determinan efecto alguno sobre el riesgo de cáncer mamario, pero dicho tratamiento está reservado solamente para aquellas pacientes histerectomizadas. En el ámbito de la TRH combinada, los estrógenos transdérmicos asociados con progesterona micronizada no parecen dar aumento significativo del riesgo aun en tratamientos de duración superior a los 4 años (1,100,101). En las pacientes que ya han sido tratadas por cáncer de mama hay contraindicación absoluta para el uso de TRH (88).Ante una IOP de etiología autoinmune, los esteroides poseen valor clínico limitado, pudiendo acarrear mayores complicaciones. Aquellas pacientes con tireopatía autoinmune y una IOP de menos de dos años de duración, pueden beneficiarse de altas dosis de glucocorticoides durante cortos períodos y obtener buenos resultados
(43,92-94).Acerca de otras contraindicaciones de la TRH podemos señalar que en el caso de las porfirias no hay datos en la literatura que establezcan relación alguna entre menopausia espontánea y porfiria. La vía transdérmica permite proseguir el tratamiento combinando los progestágenos con gel o cremas vaginales aunque no hay al momento estudios disponibles
(1). La porfiria cutánea tarda contraindica en forma absoluta la TRH (88).En aquellas pacientes con emicrania una TRH de tipo combinado continuo debe preferirse, pues en las mujeres susceptibles a las fluctuaciones de los niveles de estrógenos circulantes una TRH cíclica en posmenopausia puede exacerbar la emicrania.
Es conocido que los estrógenos poseen propiedades litiásicas y, aun cuando la literatura reporte un aumento del riesgo de colelitiasis en pacientes con TRH, ésta no está contraindicada, ni se asocia a daño hepático significativo. Es prudente escoger una vía no oral para la suministración de estrógenos en pacientes con colelitiasis aun cuando no haya datos epidemiológicos disponibles que demuestren una acción desfavorable de la vía oral
(1). Hay acuerdo general en contraindicar la TRH en pacientes con patología hepática aguda o crónica (88).Por último, el riesgo de patología tromboembólica está ligado mayormente a la presencia de anomalías congénitas o adquiridas como son el déficit de AT III, proteína C o S, anticuerpos antifosfolípidos, mutación de Leiden (mutación puntiforme del factor V del sistema de coagulación). En el caso de antecedentes de patología tromboembólica ha de preferirse la vía transdérmica para la TRH. La presencia de una patología de trombosis venosa aguda o reciente contraindican de forma absoluta toda TRH
(1,88).El tratamiento de la infertilidad en pacientes con IOP, requiere reemplazar la función ovárica tanto desde el punto de vista hormonal como de la gametogénesis
(95). Numerosos autores han reportado la reaparición de menstruaciones y más aún de embarazos, lo que indica que el cese de la actividad ovárica puede no ser irreversible y la posibilidad de un embarazo no se excluye aun cuando ésta sea remota (43).En la literatura se reporta un caso de delección parcial del brazo largo del cromosoma X 46,Xdel(X)(q22), en el cual se obtiene la inducción de la ovulación
(96). En otro caso una paciente con IOP portadora de disgenesia gonadal con mosaicismo cromosómico (86 %, 46XX; 7 % 47XXX; 7 % 45XO) logra dos embarazos; el primero posterior al tratamiento de inducción de la ovulación con gonadotropinas y el segundo con el uso de análogos del GNRH y gonadotropinas (97). Si bien los casos anteriormente reportados reflejan, que el cese de la actividad ovárica no es del todo completa, la coexistencia de una alterada calidad de los oocitos hace que estas pacientes no tengan indicación al tratamiento para la inducción de la ovulación, y la mayoría de los embarazos se presentan después de TRH (1,8,14,67,90,93,94,98,99). En 2006 Bufalino y col. (23), describen el caso de una paciente de 22 años con diagnóstico de mosaicismo de Turner en tratamiento con terapia hormonal en forma cíclica quien al consultar por amenorrea se le realiza ecografía que reportó embarazo simple intrauterino (23).Los tratamientos actualmente disponibles para preservar la fertilidad son; la donación de ovocitos o embriones; la maduración in vitro de ovocitos (M.I.O); la transferencia de citoplasma
(1,19,29,43, 55,65,91,99,102-104).En el caso de la donación de ovocitos o de embriones, la selección de la donante y la preparación endometrial de la receptora han de ser muy bien evaluadas. La edad y la historia de la paciente, deben conducir a la aplicación de un adecuado tratamiento.
La técnica prevee el uso de protocolos de estimulación de la superovulación en la paciente donante y la preparación endometrial de la receptora. Las indicaciones más comunes están representadas por: síndrome del ovario resistente, las disgenesias gonadales con cariotipo normal o anormal, y en aquellas enfermedades genéticas transmitidas sexualmente
(1,29,43,79,105-107).La maduración in vitro de oocitos (M.I.O) es el proceso, a través del cual, los oocitos inmaduros (estadio de profase l) obtenidos por aspiración de los pequeños folículos antrales de los ovarios no estimulados, maduran alcanzando el estadio de metafase II.
Estos oocitos pueden ser inseminados y continuar todas las etapas de un común ciclo de reproducción asistida. Actualmente representa aún, una técnica experimental (1,29,43,81). Los progresos en el conocimiento de las causas moleculares de la IOP y la identificación de marcadores ("target") autoinmunes en el ovario, aunado al conocimiento de que la mayoría de las pacientes con IOP poseen folículos ováricos a la ecografía, hacen que esta técnica represente un acontecimiento terapéutico en el tratamiento alternativo de pacientes con IOP, si bien es obligatoria la cautela en orden a esta técnica, en base a estudios hechos sobre ratones que evidencian una incidencia 2-3 veces mayor a la normal, de cigotes con aneuploidías y malformaciones fetales después de la crioconservación del ovocito. Puede representar una alternativa terapéutica válida para pacientes con historia familiar de IOP y para aquellas que deberán someterse a radioterapia o quimioterapia (1,43,79,108,109).La transferencia de citoplasma es una técnica de muy reciente aparición y cuyo fundamento se basa en transferir el citoplasma de mujeres jóvenes infundiéndolo en los oocitos de mujeres con edad avanzada para poder así mejorar las potencialidades de desarrollo de los mismos. Tal técnica se encuentra en la actualidad en estado experimental
(1).Los tratamientos anteriormente señalados plantean interrogantes de orden ético y moral. La edad materna avanzada, la adecuada selección de las donantes, la doble maternidad: la biológica (de la donante) y la afectiva (la de la receptora) son problemas que con seguridad ameritarán normas legislativas muy precisas (1,110).
REFERENCIAS
1. D Ambrogio G, Cela V. Menopausia e Infertilitá. En: Genazzani A.R, Gambacciani M, editores. Premenopausa e Menopausa. Fisiopatologia, clínica e terapia. Roma: CIC Edizioni Internazionali; 2000.p.29-39. [ Links ]
2. Touraine Ph. Insufficienza ovárica primitiva. Giorn It Ost. 2000;XXII(7/8):315. [ Links ]
3. Cibula D, Zivny J. Premature ovarian failure syndrome. Ceska Gynekol. 2000;65:98-102. [ Links ]
4. Christin–Maitre S. Premature ovarian insufficiency. Rev Prat. 1999;49:1297-1302. [ Links ]
5. Agüero O. Estudios sobre climaterio y menopausia en Venezuela. Rev Obstet Gynecol Venez. 1997;57:125-132. [ Links ]
6. Barbarino-Monnier P. Premature ovarian failure. J Gynecol Obstet Biol Reprod. 2000;29:316-318. [ Links ]
7. Vegetti W, Tibiletti MG, Testa G, De Lauretis Yankowski L, Alagna F, et al. Inheritance in idiopathic premature ovarian failure: Analysis of 71 cases. Hum Reprod. 1998;13:1796-1800. [ Links ]
8. Barbarino-Monnier P. [From pathological diagnosis to ovulation induction]. The case of ovarian isufficiency. Gynecol Obstet Fertil. 2001;29:39-48. [ Links ]
9. Coulam CB, Stern JJ. Immunology of ovarian failure. Am J Reprod Immunol. 1991;25:169-174. [ Links ]
10. Yabur JA. Menopausia. En: Terán Dávila J, Febres Balestrini F, editores. Endocrinología Ginecológica y Reproducción Humana. Caracas: Editorial Ateproca; 1995.p.235-289. [ Links ]
11. Tan SL, Hague WM, Becker F, Jacobs HS. Autoimmune premature ovarian failure with polyendocrinopathy and spontaneous recovery of ovarian follicular activity. Fertil Steril. 1986;45:421-424. [ Links ]
12. Terán Dávila J, Rosales JC. Menopausia prematura: ¿Una entidad irreversible? Rev Obstet Ginecol Venez. 1997;57:57-60. [ Links ]
13. Kalantaridou SN, Nelson LM. Premature ovarian failure is not premature menopause. Ann NY Acad Sci. 2000;900:393-402. [ Links ]
14. Causio F, Fischetto R, Leonetti T, Shonaver L. Ovarian stimulation in a woman with premature ovarian failure and X-autosome translocation: A case report. Repro Med. 2000;45:235-239. [ Links ]
15. Bondy CA, Nelson LM, Kalantaridou SN. The genetic origins of ovarian failure. J Womens Health. 1998;7:1225-1229. [ Links ]
16. Taylor AE. Sistemic adversities of ovarian failure. J Soc Gynecol Invest. 2001;8(Suppl 1):7-9. [ Links ]
17. Davis SR. Premature ovarian failure. Maturitas. 1996;23:1-8. [ Links ]
18. Anasti JN. Premature ovarian failure: An update. Fertil Steril. 1998;70:1-15. [ Links ]
19. Kalantaridou SN, Davis SR, Nelson LM. Premature ovarian failure. Endocrinol Metab Clin North Am. 1998;27:989-1006. [ Links ]
20. Gossain VV, Carella MJ, Rovner DR. Pregnancy in a patient with premature ovarian failure. J Med. 1993;24:393-402. [ Links ]
21. Spera G, Martini P, Cornoldi A, Morganti A, Falcone S, Lubrano C. Endocrinologia della menopausa e del climaterio. Giorn It Ost Gin. 1998;331-333. [ Links ]
22. Carlson MB. Human embryology and developmental biology. Filadelfia: Mosby; 2004.p.411. [ Links ]
23. Bufalino G, Licha M, Arcia O, Aponte A. Mosaicismo Turner 45X0/46/XX y embarazo espontáneo. Rev
Obstet Ginecol Venez. 2006;66:33-37. [ Links ]24. Cittadini E, Gattuccio F, La Sala G, Palermo R. Anovulazione cronica da causa ovarica. En: Cittadini E, Gattuccio F, La Sala G, Palermo R, editores. La sterilita umana. Palermo: Cofese; 1990.p.151-156. [ Links ]
25. Forleo R, Sbiroli C, Di Tondo U. Differenziazione Sessuale. En: Forleo R, Sbiroli C, Di Tondo U, editores. Fisiología della riproduzione femminile Milano: Gelmini Edizioni. 1982.p.1-24. [ Links ]
26. Kably A, Barroso G. Introducción a la anatomía y fisiología de la concepción humana. En: Edwards R, Risquez F, editores. Reproducción asistida moderna. Montevideo: Ares trading Uruguay s.a.; 2003.p.76. [ Links ]
27. Christin–Maitre S, Bouchard P. Genes and ovarian insufficiency. Ann Endocrinol. 1999;60:118-122. [ Links ]
28. Sherman S. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189-194. [ Links ]
29. Terán Dávila J, Teppa A, Molina R, Febres F. Insuficiencia ovárica prematura ("menopausia precoz") En: Terán J, Febres F, editores. Medicina del climaterio y la menopausia. Caracas: Editorial Ateproca; 1999.p.193-207. [ Links ]
30. Vianna-Morgante AM, Costa SS, Pares AS, Verreschi IT. FRAXA premutation associated with premature ovarian failure. Am J Med Genet. 1996;64:373-375. [ Links ]
31. Conway GS, Payne NN, Webb J, Murray A, Jacobs PA. Fragile X premutation screening in women with premature ovarian failure. Hum Reprod. 1998;13:1184-1187. [ Links ]
32. Conway GS, Hettiarachchi S, Murray A, Jacobs PA. Fragile X premutations in familial premature ovarian failure. Lancet. 1995;(8935):309-310. [ Links ]
33. Murray A, Webb J, MacSwiney F, Shipley EL, Morton NE, Conway GS. Serum concentrations of follicle stimulating hormone may predict premature ovarian failure in FRAXA premutation women. Human Reprod. 1999;14:1217-1218. [ Links ]
34. Marozzi A, Vegetti W, Manfredini E, Tibiletti MG, Testa G, Crosignani PG, et al. Association between idiopathic premature ovarian failure and fragile X permutation. Hum Reprod. 2000;15:197-202. [ Links ]
35. Banerjee N, Kriplani A, Takkar D, Kucheria K. Balanced X;22 translocation in a patient with premature ovarian failure. Acta Genet Med gemello1 (Roma). 1997;46:241-244. [ Links ]
36. Terán Dávila J, Cadena LF, Caripidis J, Azulay M. Amenorrea primaria por translocación cromosómica del X al 20. Rev Lat Amer Est Fert. 1977;11:162-165. [ Links ]
37. Center JR, McElduff A, Roberts CG. Premature ovarian failure and ovarian dysgenesis associated with balanced and unbalanced X-6 translocations, respectively: Implications for the investigation of ovarian failure. Aust NZJ Obstet Gynaecol. 1994;34:185-188. [ Links ]
38. Bione S, Sala C, Manzini C, Arrigo G, Zuffardi O, Banfi S, et al. A human homologue of the Drosophila melanogaster diaphanous gene is disrupted in a patient with premature ovarian failure: Evidence for conserved function in oogenesis and implications for human sterility. Am J Hum Genet. 1998;62:533-541. [ Links ]
39. Marozzi A, Manfredini E, Tibiletti MG, Furlan D, Walter Vegetti N, Crosignani PG, et al. Molecular definition of Xq common-deleted region in patients affected by premature ovarian failure. Human Genetics. 2000;107:311-314. [ Links ]
40. Davison RM, Quilter CR, Webb J, Murray A, Fisher AM, Valentine A, et al. A familial case of X chromosome deletion ascertained by cytogenetic screening of women with premature ovarian failure. Human Reprod. 1998;13:3039-3041. [ Links ]
41. Christin-Maitre S, Vasseur C, Portnoi MF, Bouchard P. Genes and premature ovarian failure. Mol Cell Endocrinol. 1998;145:75-80. [ Links ]
42. Maneschi M, Martorana A. Amenorree. En: Maneschi M, Martorana A, editores. Ginecología Endocrinologica clinica e terapia. Roma: CIC Edizioni Internazionali; 1986.p.62-106. [ Links ]
43. Tejerizo L, Tejerizo A, Gómez M. La menopausia precoz. Revisión de conjunto. To Ko-gin pract. 2000;59:17-38. [ Links ]
44. Terán J, Febres F, Arcia O, Turmero J. Varones cariotípicamente normales con utero y fenotipo femenino (Síndrome de Swyer). Rev Obstet Ginecol Venez. 1989;49:84-87. [ Links ]
45. Scucces M, Panecassio A. Síndrome de Morris: Reporte de un caso. Rev Obstet Ginecol Venez. 2000;60:193-196. [ Links ]
46. Caraballo Mata JA, Sánchez de la Cruz B, Perera Pérez AJ, Carrero Cuberos FM, Arias Briceño ME, De Abreu Rodríguez L. Tres hermanas con insensibilidad periférica a los andrógenos. Rev Obstet Ginecol Venez. 2001;61(1):61-65. [ Links ]
47. Cunha A. Disgenesia gonadal. En: Sánchez de La Cruz B, editora. Ginecología Infanto Juvenil. Caracas: Ateproca; 1997.p.101-107. [ Links ]
48. Pescetto G, De Cecco L, Pecorari D, Ragni N. Manuale di Ginecologia e Ostetricia. Roma: Universo; 1989. [ Links ]
49. Terán Dávila J, Teppa Garrán A, Camargo Marín L. Lo inexplicable en el síndrome de Turner puro: su presentación con desarrollo puberal y menstruaciones normales. Rev Obstet Ginecol Venez. 2000;60:213-216. [ Links ]
50. Bufalino G, Fabrega R, Gonzalez C, Romero G. Mosaicismo Turner 45 X0/XY. Diagnóstico diferencial con disgenesia gonadal mixta. Rev Obstet Ginecol Venez. 2007;67:61-66. [ Links ]
51. Riveras G, Campos A, López J. Disgenesia gonadal mixta. Reporte de un caso y revisión de la literatura. Rev Venez Cirugía. 1987;45:166-169. [ Links ]
52. Amati P, Gasparini P, Zlotogora J, Bonneau D. A gene for premature ovarian failure associated with eyelid malformation maps to chromosome 3q 22-q23. Am J Hum genet. 1996;58:1089-1092. [ Links ]
53. Kreuser ED. Ovarian insufficiency induced by cytotoxic drugs. Internist. 2000;41:67-70. [ Links ]
54. Richard J S. Molecular loci for potencial drug toxicity in ovaries. Environ Health Perspect. 1986;70:159-161. [ Links ]
55. Meirot D. Ovarian injury and modern options to preserve fertility in female cancer patients treated with high dose radio-chemotherapy for hemato-oncological neoplasias and other cancers. Leuk Lymphoma. 1999;33:65-76. [ Links ]
56. Falsetti L, Scalchi S, Villani MT, Bugari G. Premature ovarian failure. Gynecol Endocrinol. 1999;13:189-195. [ Links ]
57. Coulam CB, Ryan RJ. Prevalence of circulating antibodies directed toward ovaries among women with premature ovarian failure. Am J Reprod Immunol Microbiol. 1985;9:23-24. [ Links ]
58. Weatcroft NJ, Toogood AA, Li TC, Cooke I D, Weetman AP. Detection of antibodies to ovarian antigens in women with premature ovarian failure. Clin Exp Immunol. 1994;96:122-128. [ Links ]
59. Chattopadhyay D, Sen MR, Katiyar P, Pandey LK. Antiovarian antibody in premature ovarian failure. Indian J Med Sci. 1999;53:254-258. [ Links ]
60. Kirsop R, Brock CR, Robinson BG, Baber RJ, Wells JV, Saunders DM. Detection of anti-ovarian antibodies by indirect immunoflurescence in patients with premature ovarian failure. Reprod Fertil Dev. 1991;3:537-541. [ Links ]
61. Degros V, Pons L, Ghulam A, Racadot A. [21-hidroxylase auantibodies in patients with autoimmune endocrinopathies]. Ann Biol Clin. 1999;57:705-709. [ Links ]
62. Myhre AG, Halonen M, Eskelin P, Ekwall O, Hedstrand H, Rorsman F, et al. Autoimmune polyendocrine syndrome type 1 (APS 1) in Norway. Clin Endocrinol (Oxf). 2001;54:211-217. [ Links ]
63. Weacker P, Emmerich D, Runge M, Breckwoldt M. [Primari ovarian insufficiency in polyendocrinopathy syndrome]. Geburtshilfe Frauenheilkd. 1991;51:1004-1005. [ Links ]
64. Russel P. The Clinocopathological features of primary ovarian. Verh Dtsch Ges Pathol. 1997;81:197-209. [ Links ]
65. Kim JG, Moon SY, Chang YS, Lee JY. Autoimmune premature ovarian failure. J Obstet Gynaecol. 1995;21:59-66. [ Links ]
66. Weatcroft N, Weetman AP. Is premature ovarian failure an autoimmune disease? Autoimmunity. 1997;25:157-165. [ Links ]
67. Smitth S, Hosid S. Premature ovarian failure associated with autoantibodies to the zona pellucida. Int J Fertil Menopausal Stud. 1994;39:316-319. [ Links ]
68. Ishizuka B, Kuday Y, Amemiya A, Yamada H, Matsuda T, Agata T. Antiuclear antibodies in patients with premature ovarian failure. Human Reprod. 1999;14:70-75. [ Links ]
69. Kinugawa C, Murakami T, Okamura K, Yajima A. Telomerasa activity in normal ovaries and premature ovarian failure. Toko J Exp Med. 2000;190:231-
238. [ Links ]70. Nelson L M, Anasti JN, Kimzey LM, Defensor RA, Lipetz KJ, White BJ, et al. Development of luteinized graafian follicles in patients with karyotypically normal spontaneous premature ovarian failure. J Clin Endocrinol Metab. 1994;79:1470-1475. [ Links ]
71. Takahashi K, Ozaki T, Okada M, Kurioka H, Kanasaki H, Miyazaki K. Increased prevalence of luteinizing hormone beta-subunit variant in patients with premature ovarian failure. Fertil Steril. 1999;71:96-101. [ Links ]
72. van Kasteren YM, Hundscheid R, Snits AP, Cremers FP, Van Zonnereld P, Braat DD. Familial idiopathic premature ovarian failure: An overrated and underestimated genetic disease? Human Reprod. 1999;14:2455-2459. [ Links ]
73. Mattison DR, Evans MI, Schwimmer WB, White BJ, Jensen B, Schulman JD. Familial premature ovarian failure. Am J Hum Genet. 1984;36:1341-1348. [ Links ]
74. Conway GS, Kaltsas G, Patel A, Davies MC, Jacobs HS. Characterization of idiopathic premature ovarian failure. Fertil Steril. 1996;65:337-341. [ Links ]
75. Shah A, Mithal A, Bhatia E, Godbole MM. Extraovarian endocrine abnormalities in north Indian women with premature ovarian failure. Natl Med J Indian. 1995;8:9-12. [ Links ]
76. Kim TJ, Anasti JN, Flack MR, Kimzey LM, Defensor RA, Nelson LM. Routine endocrine screening for patients with karyotypically normal spontaneous premature ovarian failure. Obstet Gynecol. 1997;89:777-779. [ Links ]
77. Coulam CB, Ryan RJ. Prevalences of circulating antibodies directed toward ovaries among woman: With premature ovarian failure. Am J Reprod Immunol Microbiol. 1985;9:23-24. [ Links ]
78. Fenichel P, Sosset C. [Autoimmune precocious menopause: Therapeutic consequences] Contracept Fértil Sex. 1997;25:639-642. [ Links ]
79. Laml T, Schulz-Lobmeyr I, Obruca A, Huber JC, Hartmann BW. Premature ovarian failure: Etiology and prospects. Gynecol Endocrinol. 2000;14:292-
302. [ Links ]80. Metha AE, Matwijiw I, Lyons EA, Faiman C. Non invasive diagnosis of resistant ovary syndrome by ultrasonografy. Fertil Steril. 1992;57:56-61. [ Links ]
81. Conway GS. Premature ovarian failure. Curr Opin Obstet Gynecol. 1997;9:202-206. [ Links ]
82. Abache de Méndez N. Climaterio precoz. Rev Obstet Gynecol Venez. 1972;32:309-314. [ Links ]
83. Febres Balestrini F. Reemplazo hormonal oral en la post-menopausia. Rev Obstet Ginecol Venez. 1994;54:165-168. [ Links ]
84. Metka M, Heytmanek G, Enzelsberger H, Huber J, Kurz C, Spona J. Hypergonadotropic amenorrhea (WHO III) as osteoporosis risk. Geburtshilfe Frauenheilkd. 1990;50:974-976. [ Links ]
85. Bruni V, Dei M, Bigozzi L. Ipoestrogenismo adolescenziale e metabolismo osseo. Giorn It Ost Gin. 1998;(8):355-360. [ Links ]
86. Tibiletti MG, Testa G, Vegetti W, Alagna F, Taborelli M, Dalpra L, et al. The idiopathic forms of premature menopause and early menopause show the same genetic pattern. Hum Reprod. 1999;14:2731-2734. [ Links ]
87. Velásquez N, Fernández Michelena M. Efectos poco publicados de los estrógenos. Revisión. Rev Obstet Ginecol Venez. 2004;64:139-153. [ Links ]
88. Genazzani AR, Gambacciani M, Simoncini T. Menopausa e terapia ormonale sostitutiva. Giorn It Ost Ginecol. 2007;XXIX:213-222. [ Links ]
89. van Kasteren Y. Treatment concepts for premature ovarian failure. J Soc Gynecol Investig. 2001;8(1Suppl Related Articles Proceedings):58-59. [ Links ]
90. Gaddducci A, Negri S, Muraca S, Genazzani AR. Terapia ormonale sostitutiva dopo tumori non ginecologici, meningiomi, tumori emopoietici e tumori del colon. Gior It Ost Gin. 2001;XXIII:489-494. [ Links ]
91. Triantafilo Y, Arteaga E, Duque G, Fernández C, Grege G. Pregnancy in a patient with premature ovarian failure secondary to chemotherapy. Rev Med Chil. 1991;119:60-63. [ Links ]
92. Corenblum B, Rowet T, Taylor PJ. High-dose, short term glucocorticoids for the treatment of infertility resulting from premature ovarian failure. Fertil Steril. 1993;59(5):988-991. [ Links ]
93. Borini A, Bafaro G, Violini F, Flamigni C. Pregnancies in postmenopausal program. Fertil Steril. 1995;63:258-261. [ Links ]
94. Kalantaridou SN, Braddock DT, Patronas NJ, Nelson
LM. Treatment of autoimmune premature ovarian failure. Human Reprod. 1999;14:1777-1782. [ Links ]95. Weeler CA. Premature ovarian failure: Treatment strategies. RI. Med. 1995;78:130-131. [ Links ]
96. Ishizuka B, Kudo Y, Amemiya A, Ogata T. Ovulation induction in a woman with premature ovarian failure resulting from a partial deletion of the X chromosome long arm 46 X,del (X)q22. Fertil Steril. 1997;68:931-934. [ Links ]
97. Nawroth F, Foth D, Stute P, Schmidt, Romer T. Posibilities of sterility therapy in a patient with a premature menopause due to an x- chromosomal anomaly a case report. Maturitas. 2000;37:129-132. [ Links ]
98. Check JH, Nowroozi K, Nazari A. Viable pregnancy in a woman with premature ovarian failure treated with gonadotropin suppression and human menopausal gonadotropin stimulation. A case report. J Reprod Med. 1191;36:195-197. [ Links ]
99. Gucer F, Urdl W, Pieber D, Arikan MG, Giulani A, Auner H. Pregnancies in patients with premature ovarian failure. Clin Exp Obstet Gynecol. 1997;24:130-132. [ Links ]
100. López Herrera L, Alvarado H, Suárez Chacón R. Ausencia de correlación entre el hipoestrogenismo y los receptores estrogénicos en pacientes postmenopáusicas con carcinoma mamario. Acta Oncol Venez. 1985;18:59-68. [ Links ]
101. Biglia N, Kubatzki F, Ujcic E, Wandji O, Marenco D, Sgandurra P, et al. HRT e mammella: Influenza dei progestinici. Giorn It Ost Gin. 2005;XXVII(12/12):517-519. [ Links ]
102. Van Kasteren YM. Premature ovarian failure. Ned Tijdschr Geneeskd. 2000;144:2380-2383. [ Links ]
103. Van Kasteren YM, Schoemaker J. Premature ovarian failure: A systematic review on therapeutic interventions to restore ovarian function and achieve pregnancy. Hum Reprod Update. 1999;5:483-492. [ Links ]
104. Fiedler K, Wurfel W, Krusmann G, Rothenaicher M, Hirsch P, Krusmann W Sr. Course of pregnancy and labor following in vitro fertilization. A retrospective study of 246 deliveries. Z Geburtshilfe Perinatol. 1990;194:8-12. [ Links ]
105. Andersen AN, Larsen JF, Hornnes PJ, Starup J, Andersen CY, Westergaard LG, et al. [Ovum donation. A review of and a suggestion to unified guidelines for treatment at public fertility clinics in Denmark] Ugeskr Laeger. 1993;155:2515-2519. [ Links ]
106. Asch R, Balmaceda A, Borrero C, Gastaldi C, Rojas F. Oocyte donation and gamete intrafallopian transfer in premature ovarian failure. Fertil Steril. 1988;49:263-266. [ Links ]
107. Mardesic T, Hulvert J, Mikova M, Müller P, Voboril J, Huttelova R, et al. [Hormone replacement for pregnancy in functionally agonadal women after transfer of thawed embryos in the in vitro fertilization program-problems of placental incompetence]. Ceska Gynekol. 1998;63:402-405. [ Links ]
108. Ferraretti AP, Gianaroli L, Magli C, Gergolet M, Andersier C. Futuri sviluppi della maturazione in vitro degli oociti. Giorn It Ost Gin. 1999;XXI:536-537. [ Links ]
109. Rosales JC, Prado A, Camejo MI. Oogénesis y folículogénesis. En: Pagés G, Aller J, editores. Infertilidad. Fisiopatología, diagnóstico y tratamiento. Caracas: AMOLCA; 2006.p.35-58. [ Links ]
110. García de Yegüez M. Concepción ética en salud reproductiva. Rev Obstet Ginecol Venez. 2007;67:55-60. [ Links ]

