










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Obstetricia y Ginecología de Venezuela
versión impresa ISSN 0048-7732
Rev Obstet Ginecol Venez vol.76 no.2 Caracas jun. 2016
Trastornos de diferenciación sexual
Dras. Rita Pizzi1, Leticia Parpacén1, Jessica Fernandes1, Evelyn Hernández 1, Liliana Fung1, Indira Centeno1,2
1 Servicio de Endocrinología y Metabolismo, Unidad de Endocrinología Ginecológica, Hospital Universitario de Caracas. Universidad Central de Venezuela. Caracas, Venezuela.
2 Cátedra de Ginecología, Departamento de Obstetricia y Ginecología. Universidad Central de Venezuela. Caracas.
RESUMEN
Objetivo: Presentar la clínica, citogenética y hallazgos histopatológicos en pacientes adultas, que consultaron a la Unidad de Endocrinología Ginecológica del Hospital Universitario de Caracas con trastornos de la diferenciación sexual. Se reportan cuatro casos clínicos: dos casos con trastorno de la diferenciación sexual 46, XY por alteración en la acción de los andrógenos anteriormente denominado insensibilidad androgénica parcial, una paciente con trastorno de la diferenciación sexual 46, XX y otra con trastorno de la diferenciación sexual 46, XY ovotesticular sin gonadoblastoma por síndrome de Frasier. Es importante realizar un diagnóstico temprano para su tratamiento precoz, por la trascendencia que la definición del sexo tiene para el futuro del individuo.
Conclusiones: A pesar de los avances alcanzados a lo largo de los últimos 20 años, algunos casos quedan aún sin diagnóstico etiológico definido, sea por falta de estudio molecular o genes aún no conocidos. Su abordaje diagnóstico y terapéutico es complejo, requiere de un equipo multidiscplinario integrado por ginecólogos, endocrinólogos, psiquiatras, urólogos, cirujanos plásticos.
Palabras clave: Trastornos de la diferenciación sexual. Amenorrea primaria. Genitales ambiguos. Síndrome de insensibilidad androgénica.
SUMMARY
The aim of this paper is to present the clinical, cytogenetic and histopathological findings in adult patients who consulted the Gynecological Endocrinology Unit of the University Hospital of Caracas with Disorders of sexual development. Four clinical cases reported: Two with Disorder of sexual development 46, XY due defect in androgen action previously called partial androgen insensitivity, one patient with disorders of sexual development 46, XX and another with disorder of sexual development 46, XY ovotesticular without gonadoblastoma by Frasier syndrome. It is important an early diagnosis and treatment to define the sex for the individuals future.
Conclusion: Despite the progress made over the last 20 years, some cases are still without etiologic diagnosis, either through lack of molecular study or yet unknown genes. Its diagnostic and therapeutic approach is complex, requiring a team of gynecologists, endocrinologists, psychiatrists, urologists, plastic surgeons.
Key words: Disorders of sexual development. Primary amenorrhea. Ambiguous genitalia. Androgen insensitivity syndrome.
INTRODUCCIÓN
La asignación del sexo no siempre es un acto sencillo. De hecho, el sexo puede ser definido de varias maneras (1):
-. Sexo genético: definido como la constitución cromosómica del individuo, típicamente los hombres son 46, XY y las hembras 46, XX.
-. Sexo fenotípico: definido por las características de los caracteres sexuales del individuo, generalmente son femeninas o masculinas pero a veces pueden ser ambiguas o estados intersexuales.
-. Sexo gonadal: que se corresponde con el tipo de gónada que posee el individuo, generalmente testículos u ovarios pero que en raras circunstancias pueden estar ambas gónadas presentes en un individuo.
La determinación del sexo depende del sexo cromosómico en el embrión, el cual está establecido por múltiples eventos moleculares que producen directamente el desarrollo de las células germinales, su migración del seno urogenital y la formación de testículos en presencia del cromosoma Y (46, XY), o de ovarios en ausencia del cromosoma Y con la presencia del segundo cromosoma X (46, XX) (2).
El sexo está genéticamente determinado durante la fertilización por la presencia o ausencia del cromosoma Y, un gen de este cromosoma, región determinante del sexo del cromosoma Y (SRY) es el responsable de los procesos de transcripción y la diferenciación de la gónada bipotencial en testículos.
Los diferentes estadios de la diferenciación sexual dependerán de las respuestas específicas de los tejidos a las hormonas producidas por las gónadas lo que conllevará a la diferenciación del patrón masculino o femenino.
Las alteraciones de la diferenciación sexual, se refieren a fenotipos en los cuales el sexo específico es difícil de asignar, debido a su gran variabilidad que engloba niveles diversos de diferenciación, que pueden ir desde aspecto femenino en un individuo XY pasando por los genitales ambiguos, hasta el hombre infértil. La causa de estos problemas es la alteración de la compleja red de regulación de genes responsables para el desarrollo adecuado de los testículos u ovarios en el embrión (2).
Los trastornos de diferenciación sexual (TDS) son afecciones congénitas que se caracterizan por el desarrollo atípico del sexo cromosómico, gonadal o fenotípico. La incidencia de los TDS oscila entre 1/ 1.000 a 1/ 4 200 recién nacidos (1). La nomenclatura y clasificación y actualmente utilizada es la publicada en la Posición de Consenso de los trastornos del desarrollo sexual del año 2006, como se puede apreciar en los cuadros 1 y 2 (3).


La principal causa de los TDS es la hiperplasia suprarrenal congénita (HSC), entre otras etiologías están los síndromes de insensibilidad androgénica parcial, las disgenesias gonadales, los trastornos ovotesticulares y el exceso de andrógenos feto placentario y materno, en el Cuadro 1 podemos apreciar las diversas etiologías.
La incidencia de TDS en mujeres adultas es desconocida, se han reportado pocos casos en la literatura mundial.
El objetivo es presentar la clínica, citogenética y hallazgos histopatológicos de cuatro pacientes adultas, que consultaron a la Unidad de Endocrinología Ginecológica del Hospital Universitario de Caracas con trastornos de la diferenciación sexual. Se reportan
Caso Clínico # 1
Paciente de 26 años de edad, quien acude a consulta de endocrinología ginecológica en el año 2000 por presentar amenorrea y anormalidad de genitales externos. Antecedentes familiares con madre hipertensa, padre y hermanos aparentemente sanos. Niega antecedentes personales médicos y quirúrgicos. Amenorrea primaria, pubarquia a los 10 años, telarquia a los 12 años. Examen físico: Fenotipo femenino. Tensión arterial (TA) 120/80 mm/Hg.; talla: 1,67 m, índice de masa corporal (IMC) 22,6 kg/m2. Vello axilar escaso. Desarrollo mamario Tanner 2. Vello pubiano ginecoide Tanner 4. Genitales externos: falo de 5 x 2 cm (Figura 1), uretra vulvar, presencia de labioescroto, no se palpan gónadas en labioescroto ni en conducto inguinal, introito vaginal visible. Vagina: Fondo de saco ciego de 2 cm. Tacto rectal: No se palpan estructuras pélvicas. Extremidades: sin alteraciones. Estudios hormonales: LH: 14,4 mUI/mL, FSH: 72 mUI/mL, estradiol: 106 pg/mL, testosterona libre: 5,48 pg/mL, 17OH-progesterona: 2,0 ng/ mL. Ecosonograma pélvico en enero 2000: Útero y ovarios no visibles, en la exploración ecográfica inguinal se evidencia en canal inguinal derecho imagen de 16,8 x 7,1 mm de ecoestructura similar a testículo y en canal inguinal izquierdo imagen de 14 x 8 mm de ecoestructura similar al testículo, con imagen ecolúcida en su interior de 10 x 13 mm con reforzamiento posterior.
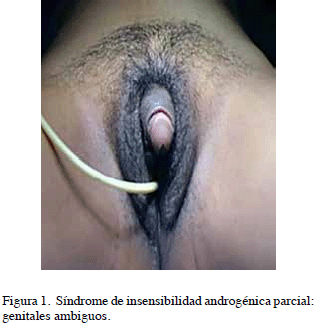
Estudio genético: Cariotipo XY. Se realizó gonadectomía, falectomía, plastia de genitales externos y neovagina con técnica de Mc Indoe – Banister. Biopsia de gónadas: tejido testicular normal. En la Figura 2 se puede apreciar la configuración actual de los genitales externos. La paciente está en terapia hormonal con estrógenos transdérmicos.
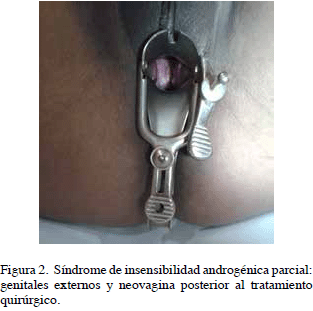
Diagnóstico: TDS 46 XY por alteración en la acción de los andrógenos, anteriormente denominado síndrome de insensibilidad androgénica parcial.
Caso Clínico # 2
Se trata de paciente de 20 años de edad, quien consultó al servicio de endocrinología ginecológica en el año 2000, por amenorrea, escaso desarrollo mamario, e inconformidad con sus genitales externos. Amenorrea primaria. Pubarquia: 12 años. Telarquia: 12 años. Fenotipo femenino. Talla: 1,61 m. IMC: 23,04. TA: 120/60 mmHg. Vello axilar presente. Vello pubiano Tanner 4, se aprecia falo de 3 cm x 1 cm. Labios mayores a predominio de los menores. Fondo de saco vaginal de 3 cm. Exámenes paraclínicos: LH: 3,3 mUI/L, FSH: 2,9 mUI/L, prolactina: 7,5 ng/mL, testosterona libre: 31,4 pg/mL, 17 OH progesterona: 2,8 ng/mL. Ultrasonido abdomino-pélvico: sin evidencia de genitales femeninos en pelvis, conductos inguinales con estructuras compatibles con gónadas Cariotipo 46 XY.
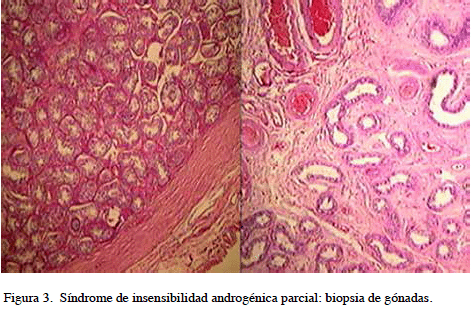
Se realizó: gonadectomía, falectomía, plastia de genitales externos y neovagina con técnica de Mc Indoe – Banister. Biopsia de gónadas: Testículos derecho e izquierdo con túbulos seminíferos sin células germinales, no observando tumores de células germinales (Figura 3). Diagnóstico: TDS 46 XY por alteración en la acción de los andrógenos, anteriormente denominado síndrome de insensibilidad androgénica parcial.
Evolución año 2015: evolución satisfactoria, en terapia hormonal con estrógenos transdérmicos. Tiene pareja sexual masculina, sin dificultad al momento del coito.
Caso Clínico # 3
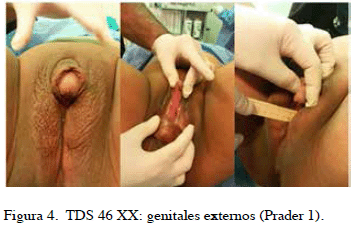
Se trata de paciente de 27 años, quien acude a la consulta de endocrinología ginecológica en 2015 por amenorrea primaria. Antecedentes familiares: 2 hermanas con amenorrea primaria y genitales ambiguos; un sobrino con diagnóstico hiperplasia suprarrenal congénita clásica. Amenorrea primaria, niega telarquia, pubarquia a los 10 años. Refiere vida sexual activa, heterosexual, sexo anal). Con libido y orgasmos presentes. Examen físico: TA: 108/68 mmHg, Talla: 1,44 m. IMC: 25,6 kg/m2. Acantosis nigricans en pliegues corporales. Implantación normal del cabello y pabellón auricular. Sin signos faciales, cervicales, ni en paladar, sugestivos de Turner. Cuello: Corto, simétrico, móvil, sin masas ni adenomegalias. Tiroides no visible ni palpable. Cardio pulmonar: sin alteraciones. Mamas: Tanner I. Distancia intermamilar aumentada. Vello axilar ausente, genitales externos ambiguos: Prader III, con senourogenital de 1 cm de profundidad. Índice clitorideo de 10 cm2 (Figura 4). Se evidencia uretra (salida de orina) hacia fondo y cara anterior de seno urogenital. Sin masas palpables en labios mayores ni regiones inguinales. Extremidades eutróficas, normotónicas. Cúbito valgus y acortamiento del cuarto y quinto metatarsiano con hallux valgus bilateral (Figura 5). Neurológico: consciente. Sin déficit neurológico. Ecosonograma pélvico: se apreció imagen alargada de 37 x 21 x 21 mm de eco textura similar al útero, sin evidencias de anexos en ambas fosas ováricas. A la exploración del conducto inguinal y zona perineal no se palpa tejidos de aspecto testicular. Ecosonograma abdominal sin alteraciones. Vulvoscopia: se evidencia mucosa pálida, con presencia de vellos al final del seno urogenital, orificio uretral en pared anterior (1/3 proximal). Estudios hormonales: TSH: 1,6 mUI/L, Anti-TPO: negativo. FSH: 0,53 mUI/L, testosterona total: 1,6 ng/dL, SHBG: 15,9 nmol/L, 17 OH progesterona: 0,9 ng/ mL. Ecocardiograma: sin alteraciones morfológicas. Urografía de eliminación: sin alteraciones. Rx ambos pies AP y Lateral: cierta desviación del eje interfalángico del 4to dedo de ambos pies. Rx de columna dorso-lumbar AP y lateral: sin alteraciones aparentes. Resonancia magnética con gadolinio y cortes milimétricos en región suprarrenal (03/02/15) sin evidencia de LOE. Diámetro suprarrenal de 11 mm (rango 10-15 mm). Cariotipo 46 XX.
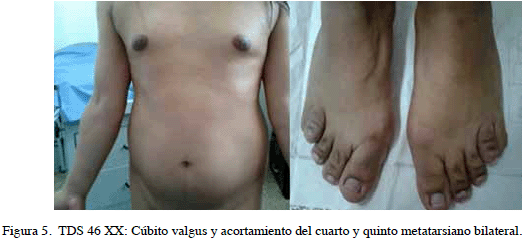
Se realizó laparotomía con hallazgos de útero y ovarios rudimentarios, se realiza clitoroplastia, ninfoplastia y neovagina por técnica de Mc Indoe. Anatomía patológica: Gónadas femeninas, sin evidencia de malignidad, endometrio hiperestrogénico, ausencia de cérvix y clítoris fibrótico. Actualmente la paciente en buenas condiciones generales, integrada socialmente con vida sexual activa. En terapia de reemplazo hormonal con 17β-estradiol.
Diagnóstico: TDS 46 XX.
Caso #4
Paciente femenina de 27 años con enfermedad renal crónica estadio V a los 19 años, en condición pos trasplante renal quien es referida a la consulta de Endocrinología por amenorrea primaria. Al examen físico se evidencia fenotipo femenino armónico, talla normal, vello púbico Tanner IV y axilar presente, mamas Tanner I, Cardiopulmonar: sin alteraciones. Abdomen: LOE en fosa ilíaca derecha, no doloroso, compatible con riñón intrapélvico y LOE en canal inguinal derecho <1 cm, no doloroso, móvil, sin hernias inguinales ni LOE en hipogastrio. Genitales externos: labios mayores de aspecto y configuración normal, Prader 1, no se palpan tumoraciones. Ecosonograma inguinal y pélvico: Riñón intrapélvico, no se observó útero ni gónadas. Cariotipo 46, XY. Estudio genético: Amplificación por PCR del ADN del gen WT1: sustitución de aminoácido C> T IVS9 + 4. Se realiza gonadectomía bilateral, cuya biopsia reportó: ovotestis bilateral sin gonadoblastoma (Figura 6)
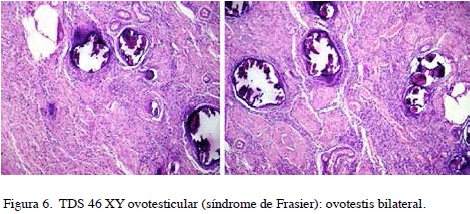
Diagnóstico: TDS 46 XY ovotesticular, síndrome de Frasier.
DISCUSIÓN
La diferenciación gonadal a testículos (1,2,4,5), está determina por un producto del gen Sex determing Region Y chromosome (SRY), presente en el brazo corto del cromosoma Y, y la proteína DAX 1 inducen la proliferación del epitelio celómico que producirán proteínas específicas como la SOX 9. Estas células que luego darán origen a las células de Sertoli, las cuales secretan la hormona anti mülleriana (HAM).
En los últimos años diversas moléculas, factores de crecimiento y factores de transcripción se han involucrado en la morfogénesis temprana de los esbozos gonadales y genitales, la mayoría involucrados en el desarrollo testicular (2,4,5). Aunque poco se conoce sobre el desarrollo del ovario, hasta el momento se ha descrito que es necesaria la presencia de factores como el SCC, c kit y Wnt4 para que ocurra esta diferenciación gonadal.
Actualmente se cree que tanto la diferenciación de los testículos como la de los ovarios requieren genes dominantes, entre ellos el SRY, que induce el desarrollo testicular mediante la regulación incremental de SOX9, además de otros genes, principalmente WNT4 y RSPO1, que se agrupan para estimar el desarrollo de los ovarios mediante la represión de dicha proteína. Este nuevo concepto considera que el destino final de la gónada bipotencial depende del equilibrio entre fuerzas opuestas y el SRY como factor clave. En las gónadas XY, el SRY induce SOX9 y orienta la diferenciación hacia el desarrollo de testículos, mientras que en las gónadas XX que carecen de SRY se combinan otros genes para reprimir SOX9 y estimular el desarrollo ovárico. En conjunto, estas observaciones son un claro indicio de que el desarrollo ovárico es el resultado de la represión activa de uno o más genes en la trayectoria testicular, y no de un mecanismo de desarrollo predeterminado.
Bajo la influencia de la testosterona, producida a partir de la semana 8 por las células de Leydig, los conductos mesonéfricos o de Wolf se diferenciaran en vasos deferentes, vesículas seminales y epidídimo. La presencia de la HAM, secretada por el testículo desde la semana 8 de gestación inhibe el desarrollo de los conductos de Müller en el feto masculino. Los conductos paramesonéfricos o de Müller formaran el útero, las trompas de Falopio y el tercio superior de la vagina en ausencia de testosterona y HAM.
Los órganos genitales externos (5,6) se originan a partir de derivados de la cloaca y la membrana cloacal. El tabique uro-rectal divide a la cloaca en dos compartimientos: el seno urogenital ventralmente, y el recto, dorsalmente. El seno urogenital interviene en la formación de la vejiga, uretra, vagina y la próstata. La membrana urogenital evoluciona formando los pliegues genitales, bordeados externamente por los repliegues labioescrotales; en el extremo anterior se forma una estructura llamada tubérculo. Los esbozos de los genitales externos son bipotenciales, su evolución en sentido masculino o femenino dependen de la presencia o ausencia de hormonas testiculares.
Los defectos en la síntesis de testosterona (5), por mutaciones en las enzimas responsables de la conversión del colesterol (Star), en la P450 17 hidroxilasa, 3 β dehidrogenasa y 17 cetoesteroide reductasa pueden causar niveles insuficientes de andrógenos; las mutaciones del gen tipo 2 de 5 α reductasa resultan en alteraciones de la diferenciación sexual.
En las mujeres, la exposición anormal a los andrógenos entre las 9 y 14 semanas de la gestación provoca distintos grados de masculinización, como la hipertrofia del clítoris y la fusión labial. En los hombres, sino se logra una actividad suficiente de los andrógenos durante el mismo intervalo crítico se produce la masculinización incompleta de los genitales, lo que genera un falo pequeño, hipospadia o defectos en los escrotos. En ambos sexos, dado que los genitales externos comparten un origen común, las ambigüedades que se observan en ellos provienen de las anormalidades en la actividad de los andrógenos, que en el caso de las mujeres es excesiva y en el de los hombres insuficientes (7).
El síndrome de insensibilidad a los andrógenos (SIA) es una enfermedad que se caracteriza por la resistencia de los tejidos diana a los andrógenos, en individuos con cariotipo 46, XY. La presentación fenotípica de SIA es variable, estos individuos generalmente desarrollan fenotipo femenino. De hecho, el SIA inicialmente fue llamado síndrome de feminización testicular porque se presentaba como mujeres aparentemente normales con un cariotipo masculino, amenorrea primaria, y presencia de testículos (8). Más tarde, se determinó que SIA era causado por anomalías en el gen del receptor de andrógenos (RA). Actualmente, se prefiere el término SIA al de feminización testicular debido a que los pacientes y los familiares se sienten más cómodos con él y porque con mayor precisión describe la fisiopatología de la enfermedad (9).
El SIA es una enfermedad poco frecuente, y su prevalencia se estima entre 1 en 90 000 a 100 000 personas. Los datos sobre la prevalencia del SIA completo indican que 1 de cada 20 000 a 64 000 de varones recién nacido se ven afectados (10), y la prevalencia de SIA parcial es desconocida (11).
El SIA parcial es menos frecuente que el SIA completo y tiene presentaciones clínicas variables que van desde feminización casi completa a casi masculinización normal, o individuos con ambigüedad sexual manifiesta (11), como los dos casos que presentamos.
Esta variabilidad refleja las diferentes mutaciones del gen RA; las formas más ambiguas son diagnosticados en el nacimiento o durante la infancia. Los pacientes con SIA parcial que se caracterizan como mujeres presentan amenorrea primaria, clitoromegalia y una fusión de los labios durante la pubertad (11).
El SIA es una enfermedad genética recesiva ligada al cromosoma X que es el resultado de mutaciones en el gen RA (Xq11-q12). No se caracteriza por deficiencias enzimáticas o la ausencia de andrógenos o de HAM. Las deleciones del gen RA han sido detectadas solo en pacientes con SIA completo. Las sustituciones de aminoácidos (típicamente en el dominio de unión a hormonas) están presentes en todas las formas de la enfermedad. En algunos casos, las mutaciones no han sido detectadas en el gen del receptor de andrógenos, lo que sugiere alteraciones pos-receptor o mosaicismo (12-15).
El diagnóstico definitivo de SIA parcial depende del momento de la consulta, si la consulta se solicita debido a la ambigüedad sexual después del nacimiento, se debe realizar un análisis de cariotipo y se confirmará con la biopsia gonadal la presencia de tejido testicular.
El perfil hormonal al nacer muestra niveles de testosterona y hormona luteínica (LH) altos o ligeramente por encima del rango normal para varones. Durante la pubertad, los individuos mantienen testosterona ligeramente elevada. La actividad del estrógeno se incrementa y actividad de la aromatasa se conserva, siendo responsable de desarrollo mamario. La HAM permanece normal porque la secreción y la función de las células Sertoli no se ve afectada (11,16).
Los pacientes con SIA tienen un mayor riesgo de desarrollar neoplasia gonadal, principalmente tumores de células germinales (gonadoblastoma y disgerminomas) (17). El riesgo es bajo antes de la edad de 25 años y es más frecuente entre 30-50 años de edad, un promedio estimado de 5 % de los pacientes desarrollan neoplasia gonadal (18-20). Por tanto, la mayoría de las directrices recomiendan posponer la gonadectomía hasta que las características sexuales estén totalmente desarrolladas o por lo menos hasta que el paciente ha alcanzado 16 años de edad. Para las pacientes con SIA parcial que son criadas como mujeres, una extirpación temprana de los testículos es preferible para evitar el crecimiento pos-puberal del clítoris y la fusión de los labios (21). En este caso, la terapia de reemplazo hormonal con estrógenos será necesaria para iniciar la pubertad e iniciar el desarrollo de rasgos femeninos y prevenir la osteoporosis (11).
Las anomalías de la diferenciación sexual con cariotipo 46, XX incluye todas las posibles anomalías de diferenciación de la gónada femenina, el ovario; entre ellas la disgenesia gonadal 46, XX, la quimera ovotesticular 46, XX y el desarrollo testicular con cariotipo 46, XX (por ejemplo, SRY+, dup SOX9, mutaciones en RSPO1). También las causas de virilización de los genitales femeninos, cuando la gónada presenta una diferenciación normal en ovario; como las hiperplasias suprarrenales congénitas, déficit de aromatasa y déficit de oxido-reductasa placentario, tumores maternos virilizantes, uso de fármacos con actividad androgénica durante el embarazo y también malformaciones urogenitales múltiples sin etiología hormonal que simulan una virilización (6,22).
En la Figura 7 se muestra un esquema para la evaluación de pacientes con TDS 46 XX, siendo el primer estudio a solicitar los niveles de 17OH progesterona sérica, debido a que la hiperplasia suprarrenal congénita constituye la primera causa de estas alteraciones (6). Actualmente está bien establecido que la etiología de un número importante de TDS son mutaciones en genes necesarios para la diferenciación sexual normal. La demostración de la mutación o mutaciones establece el diagnóstico etiológico definitivo y permite los estudios familiares, el consejo genético y el diagnóstico prenatal (22).
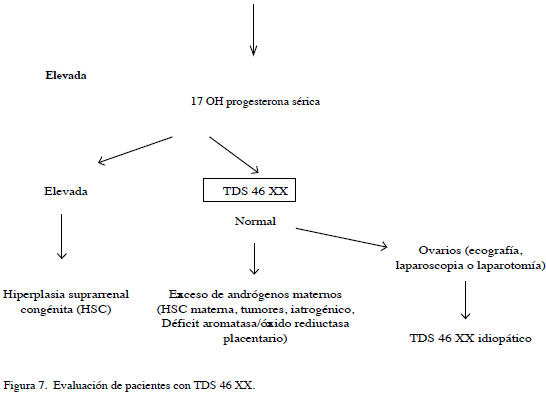
Las mutaciones detectadas pueden ir desde la deleción total del gen, que a veces incluye material genético contiguo, pasando por deleciones parciales (desde varios exones hasta una sola base), hasta cambios puntuales de un solo nucleótido que puede que puede provocar desde una parada en la pauta de lectura hasta una proteína con estructura anómala y pérdida de función (22). En el caso que presentamos, no encontramos alteraciones en los niveles de 17 OH progesterona, ni evidencias de excesos de andrógenos maternos, por tanto hasta no tener disponibles otros estudios genéticos, la paciente estaría en el grupo de TDS46 XX idiopático.
El síndrome de Frasier (23,24) es un trastorno poco frecuente, que está incluido en el grupo de TDS 46, XY y se caracteriza clínicamente por disgenesia gonadal completa, que se manifiesta por pubertad retrasada y/o amenorrea primaria, asociado a glomerulopatía rápidamente progresiva, por síndrome nefrótico, en la segunda década de la vida, que no responde a glucocorticoides y terapia inmunosupresora, que evoluciona a insuficiencia renal crónica terminal. Es causada por mutaciones específicas en el gen WT1 (Tumor de Wilms) ubicado en el cromosoma 11p23 (25). El gen WT1, está involucrado en el desarrollo gonadal y renal, así como el mantenimiento de la integridad funcional y estructural de estos tejidos. Las mutaciones que comprometen a este gen, también involucran un riesgo elevado de desarrollar tumores (25).
Se establece que este grupo de pacientes tienen alto riesgo para el desarrollo de gonadoblastomas, que puede estar presente en el 60 % de los casos (23).
Han sido reportados 88 casos en la literatura (26), sin antecedentes de casos descritos en Venezuela.
Las consecuencias psicológicas y sociales de asignación de género y las relativas al tratamiento en los pacientes con TDS, son muy importantes y requiere un enfoque multidisciplinario con inclusión de genetistas, neonatólogos, endocrinólogos, ginecólogos, psiquiatras, cirujanos. Los miembros de este multidisciplinario deben tener un especial interés en los TDS y poseer suficiente experiencia con este grupo de pacientes.
El actual debate sobre el manejo de pacientes con intersexualidad y condiciones relacionadas se centra en cuatro temas principales, a saber, diagnóstico etiológico, la asignación de género, las indicaciones y el momento de la cirugía genital, y la información para el paciente.
REFERENCIAS
1. Dewing P, Bernard P, Vilain E. Disorders of gonadal development. Sem Reprod Med. 2002;20:189-197. [ Links ]
2. MacLaughlin D, Donahoe P. Sex determination and differentiation. NEJM. 2004;350:367-378. [ Links ]
3. Hughes IA, Houk C, Ahmed SF, Lee PA; LWPES Consensus Group; ESPE Consensus Group Group. Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554-563. [ Links ]
4. Rey R. Fetal sex differentiation: From molecules to anatomy. Rev Chil Anat. 2001;19:1-14. [ Links ]
5. Cotinot C, Pailhoux E, Jaubert F, Fellous M. Molecular genetics of sex determination. Sem Reprod Med. 2002;20(3):157-167. [ Links ]
6. Öçal G. Current Concepts in Disorders of Sexual Development. J Clin Res Ped Endo. 2011;3(3):105- 114. [ Links ]
7. Sultan C. Disorders of androgen action. Semin Reprod Med. 2002;20(3):217- 227. [ Links ]
8. Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol. 1953;65:1192-1211. [ Links ]
9. Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, French FS, et al. Androgen receptor locus on the human X chromosome: Regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet. 1989;44:264-269. [ Links ]
10. Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, Rowland J, et al. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000;85:658- 665. [ Links ]
11. Mendoza N, Motos M. Androgen insensitivity síndrome. Gynecol Endocrinol. 2013;29(1):1-5. [ Links ]
12. Brüggenwirth HT, Boehmer AL, Ramnarain S, Verleun-Mooijman MC, Satijn DP, Trapman J, et al. Molecular analysis of the androgen-receptor gene in a family with receptor positive partial androgen insensitivity: An unusual type of intronic mutation. Am J Hum Genet. 1997;61:1067-1077. [ Links ]
13. Holterhus PM, Brüggenwirth HT, Hiort O, Kleinkauf-Houcken A, Kruse K, Sinnecker GH, et al. Mosaicism due to a somatic mutation of the androgen receptor gene determines phenotype in androgen insensitivity syndrome. J Clin Endocrinol Metab. 1997;82:3584- 3589. [ Links ]
14. Grino PB, Griffin JE, Cushard WG Jr, Wilson JD. A mutation of the androgen receptor associated with partial androgen resistance, familial gynecomastia, and fertility. J Clin Endocrinol Metab. 1988;66:754-761. [ Links ]
15. Köhler B, Lumbroso S, Leger J, Audran F, Grau ES, Kurtz F, et al. Androgen insensitivity syndrome: Somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling. J Clin Endocrinol Metab. 2005;90:106-111. [ Links ]
16. Boyar RM, Moore RJ, Rosner W, Aiman J, Chipman J, Madden JD, et al. Studies of gonadotropin-gonadal dynamics in patients with androgen insensitivity. J Clin Endocrinol Metab. 1978;47:1116-1122. [ Links ]
17. Sakai N, Yamada T, Asao T, Baba M, Yoshida M, Murayama T. Bilateral testicular tumors in androgen insensitivity syndrome. Int J Urol. 2000;7:390-392. [ Links ]
18. Dimitri P, Cohen M, Wright N. Indications for familial screening and gonadectomy in patients with 46, XY gonadal dysgenesis. Int J Gynaecol Obstet. 2006;95:167-168. [ Links ]
19. Dewhurst CJ, Ferreira HP, Gillett PG. Gonadal malignancy in XY females. J Obstet Gynaecol Br Commonw. 1971;78:1077-1083. [ Links ]
20. Manuel M, Katayama PK, Jones HW Jr. The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am J Obstet Gynecol. 1976;124:293-300. [ Links ]
21. Vidal I, Gorduza DB, Haraux E, Gay CL, Chatelain P, Nicolino M, et al. Surgical options in disorders of sex development (DSD) with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24:311-324. [ Links ]
22. Audí Parera L, Gracia Bouthelier R, Castaño González L, Carrascosa Lezcano A, Barreiro Conde J, Bermúdez de la Vega JA, et al.; Grupo de Trabajo sobre Anomalías de la Diferenciación Sexual de la Sociedad Española de Endocrinología Pediátrica. Anomalías de la diferenciación sexual. Protoc Diagn Ter Pediatr. 2011;1:1-12. [ Links ]
23. Hersmus R, Van der Zwan YG, Stoop H, Bernard P, Sreenivasan R, Oosterhuis JW, et al. A 46, XY Female DSD patient with bilateral Gonadoblastoma, a Novel SRY Missense Mutation Combined with a WT1 KTS Splice-Site Mutation. Plos One. 2012;7(7). [ Links ]
24. Guaragna MS, Lutaif AC, Bittencourt VB, Piveta CS, Soardi FC, Castro LC. Frasier syndrome: Four new cases with unusual presentations. Arq Bras Endocrinol Metab. 2012;56(8):525-532. [ Links ]
25. Klamt B, Koziell A, Paulat F, Wieacker P, Scambler P, Berta P, et al. Frasier Syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 ± KTS splice isoforms. Hum Mol Genet. 1998;7(4):709-714. [ Links ]
26. Ezaki J, Hashimoto K, Asano T, Kanda S, Akioka Y, Hattori M, et al. Gonadal tumor in Frasier syndrome: A review and classification. Cancer Prev Res (Phila). 2015;8(4):271-276. [ Links ]
