











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkKasmera
versión impresa ISSN 0075-5222
Kasmera v.38 n.1 Maracaibo jun. 2010
Mecanismos de resistencia a Glicopéptidos en Staphylococcus aureus
Mechanisms of Resistance to Glycopeptides in Staphylococcus aureus
Castellano González, Maribel J.1; Perozo-Mena, Armindo J.2
1Cátedra de Bacteriología General. Escuela de Bioanálisis. LUZ 2Cátedra de Práctica Profesional de Bacteriología. Escuela de Bioanálisis. LUZ. Centro de Referencia Bacteriológica –Servicio Autónomo Hospital Universitario de Maracaibo (SAHUM). *E-mail: maribeljo@cantv.net.
Resumen
Los glicopéptidos (vancomicina y teicoplanina) constituyen una alternativa terapéutica en el tratamiento de infecciones severas por cepas de S. aureus resistentes a meticilina. Sin embargo, ya se han descrito dos mecanismos de resistencia en S. aureus: resistencia de bajo nivel, caracterizada por un engrosamiento anormal de la pared celular, presente en las cepas VISA y, resistencia de alto nivel mediada por el operón vanA, que provoca la sustitución de los residuos terminales D-ala-D-ala por D-ala-D-lac, disminuyendo su afinidad por el antibiótico. Esta revisión resume la historia de la aparición de la resistencia a glicopéptidos en S. aureus y considera los mecanismos que determinan la resistencia en estos organismos como base para comprender la necesidad y los potenciales roles de nuevos agentes de esta clase.
Palabras clave: Staphylococcus aureus, glicopéptidos, vancomicina, teicoplanina, mecanismos de resistencia.
Abstract
Glycopeptides (vancomycin and teicoplanin) are an alternative therapeutic in the treatment of severe infections by methicillin-resistant S. aureus strains. However, two resistance mechanisms of S. aureus have already been described: low-level resistance, characterized by an abnormal thickening of the cellular wall, present in the VISA strains, and high-level resistance, mediated by the vanA operon, which causes the replacement of D-ala - D-ala terminal residues by D-ala-D-lac, decreasing its affinity for the antibiotic. This review summarizes the history of the emergence of glycopeptide resistance in S. aureus and considers the mechanisms that determine the resistance in these organisms as a background for understanding the need and potential roles of new agents of this kind.
Key words: Staphylococcus aureus, glycopeptides, vancomycin, teicoplanin, resistance mechanisms.
Recibido: 05-10-09 / Aceptado: 14-06-10
Introducción
Los glicopéptidos (vancomicina y teicoplanina) constituyen las principales alternativas terapéuticas de las infecciones sistémicas por S. aureus resistente a meticilina (SAMR); sin embargo, no todas las infecciones comprometen la vida y los antibióticos orales proveen un modo alternativo de terapia, particularmente cuando se requiere la administración de terapia prolongada, como por ejemplo, en presencia de material prostésico (1).
La vancomicina ha sido la droga de elección durante los últimos 30 años para el tratamiento de las infecciones por SAMR. La aparición de cepas con susceptibilidad disminuida a este antibiótico, representa un problema clínico significativo con muy pocas opciones terapéuticas (1, 2).
La vancomicina, descubierta en 1956 y aprobada por la Administración Federal de Alimentos y Drogas (FDA) en 1958, es un antibiótico polipeptídico, relativamente grande (con un peso molecular de 1.485,70; obtenido de Nocardia orientalis (Streptomyces orientalis, Amycolaptosis orientalis). Este antibiótico no interactúa ni bloquea ninguna de las enzimas que intervienen en la síntesis del peptidoglucano, como lo hacen los b-lactámicos; sino que bloquea físicamente el sustrato más importante para la maquinaria de síntesis de la pared celular, como son los residuos D-alanina-D-alanina (DDR) del lípido precursor II. En consecuencia, inhibe el empleo del sustrato por la glicosiltransferasa (enzima de síntesis de la pared celular) para producir la cadena naciente de peptidoglucano (3, 4). Sin embargo, además de estos residuos que constituyen el blanco de la vancomicina, el peptidoglucano de la pared celular de S. aureus posee cerca de 6,0 x 106 DDRs (5) a los cuales puede unirse el antibiótico, mientras penetra la capa de peptidoglucano. Por lo tanto, los residuos de la pared celular pueden actuar como falsos sustratos para la vancomicina, haciendo a la droga ineficiente en términos de mantener concentraciones eficaces alrededor de sus blancos (6).
La teicoplanina es otro de los antibióticos glicopéptidos, descubierto en 1978, difiere de la vancomicina de varias maneras. En primer lugar, hay aminoácidos aromáticos en posiciones 1 y 3, en contraste con los aminoácidos alifáticos presentes en vancomicina. La segunda diferencia es que en lugar de un disacárido que contiene vancosamina, la teicoplanina contiene un ácido graso adjunto a la glucosamina que se une a su vez a la estructura peptídica central. Esta cadena lateral de ácidos grasos, se cree que contribuye a la actividad antimicrobiana, proporcionando un medio de anclaje de la molécula a la membrana celular bacteriana (7).
Mecanismos de Resistencia a Vancomicina
Se desconoce el mecanismo verdadero de la resistencia a la vancomicina en S. aureus. Sin embargo, se han identificado dos formas de resistencia en S. aureus: resistencia de bajo nivel (observada en las llamadas cepas VISA, S. aureus intermedias a vancomicina) y de alto nivel (presente en cepas vancomicina resistentes (SAVR) (8, 9).
Resistencia de alto nivel: Se ha identificado un transposón que porta los genes de resistencia a vancomicina en enterococos (10). En presencia de vanA, el transposón Tn1546 de los enterococos puede reemplazar el DDR del peptidoglucano por D-ala-D-lactato, previniendo así, la unión a la vancomicina (11).
Desde la aparición de resistencia a vancomicina en enterococos en 1988 y la demostración in vitro, en 1992 de que esta resistencia, debida a los genes vanA y vanB, era transmisible a otras especies bacterianas incluyendo S. aureus, la posible aparición de resistencia entre cepas SAMR ha sido motivo de preocupación (11).
En el año 2002, dos cepas altamente resistentes a vancomicina de S. aureus fueron detectadas en pacientes en USA. Dichas cepas tenían Concentraciones Inhibitorias Mínimas (CIMs) a vancomicina de 1024 y 32 mg/ml y adquirieron el transposón Tn1546 a partir de una cepa de enterococos resistente a vancomicina (EVR) coexistente (12).
Hasta la fecha, 3 cepas de SAVR (S. aureus vancomicina resistente) han sido aisladas de pacientes en USA (13). Las cepas de S. aureus son resistentes a vancomicina debido a la adquisición del operón vanA a partir de enterococos resistentes a este antimicrobiano. El operón vanA permite la síntesis de un precursor anómalo de la pared celular, cuyo extremo terminal es D-ala-D-lac en vez del D-ala-D-ala habitual. Este nuevo dipéptido reduce dramáticamente la afinidad por el antibiótico. En presencia de la droga, se sintetiza el nuevo precursor de la pared, permitiendo que continúe el ensamblaje del peptidoglucano (9) (Figura 1).
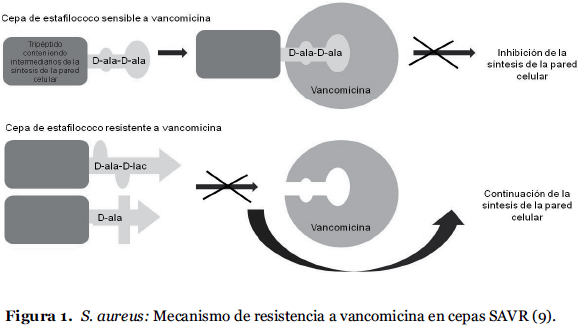
El plásmido enterocócico que contiene vanA codifica también una feromona sexual sintetizada por S. aureus, lo que representa un potencial facilitador para la transferencia por conjugación. Estos aislamientos SAVR expresan resistencia completa, con CIMs de ³ 128 µg/ml. La síntesis de D-ala-D-lac se produce sólo con la exposición a bajas concentraciones de vancomicina. Como resultado, las demandas biosintéticas adicionales son limitadas y las cepa SAVR encajan ecológicamente (9). Este ajuste ecológico, la posibilidad de que el intercambio de este plásmido se produzca más frecuentemente (debido a la creciente colonización de pacientes con SAMR y enterococos resistentes), y la resistencia de estas cepas a todos los β-lactámicos y glicopéptidos incrementa la probabilidad que las cepas SAVR se hagan rápidamente más frecuentes (3, 8, 9, 14).
Resistencia de bajo nivel: En el caso de las cepas VISA; sin embargo, ni el gen vanA ni sus homólogos han sido detectados (15, 16). En su lugar, se observó una modificación en la fisiología celular debido a la acumulación de mutaciones implicadas en su resistencia (16-18). Se han reportado alteraciones de la expresión de genes, tales como: pbp2 , pbpD (19), sigB (20, 21), ddh, icaA (22) y vraSR (23); sin embargo, ninguno de estos genes ha podido ser relacionado específicamente con la resistencia a vancomicina. Estudios realizados por microscopía electrónica demuestran que todas las cepas VISA tienen en común, un engrosamiento de la pared celular (6, 24-26), por lo tanto, existe consenso en relación a que esta característica es responsable de la resistencia a este antibiótico (6), al disminuir el coeficiente de difusión de la penetración de la droga a través de las capas de peptidoglucano de la pared celular (6); además, produce el amontonamiento de moléculas de vancomicina unidas a la pared, lo cual podría sugerir una correlación significativa entre el grosor de la pared celular y el nivel de resistencia a vancomicina (27).
La Figura 2 muestra el mecanismo de resistencia molecular propuesto para cepas VISA. Al parecer estas cepas son aisladas de poblaciones heterogéneas de cepas vancomicina resistentes. Las cepas VISA sintetizan cantidades adicionales de peptidoglucano con incremento en el número de residuos D-ala-D-ala que se unen a la vancomicina, evitando que el antibiótico alcance su sitio de acción en la pared celular (9).
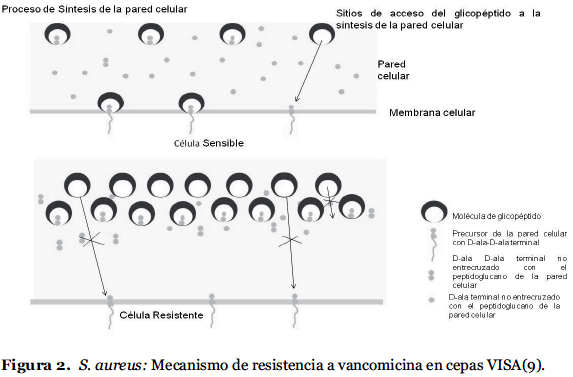
Las cepas VISA poseen una CIM a vancomicina de 8–16 µg/ml (14). También se ha detectado una etapa pre-VISA de resistencia, heterogéneamente resistente. Las cepas hetero-resistentes siguen siendo susceptibles a la vancomicina, pero contienen subpoblaciones resistentes. Los aislamientos VISA son seleccionados de las subpoblaciones resistentes debido a la exposición a vancomicina (3, 8, 9, 14). La reducida susceptibilidad al antibiótico aparece como resultado de cambios en la biosíntesis del peptidoglucano. Las cepas VISA se caracterizan por la síntesis de notables cantidades adicionales de peptidoglucano que resulta en forma irregular, en un engrosamiento de las paredes celulares. Se produce también una disminución del entrecruzamiento de las hebras de peptidoglucano, que conduce a la exposición de más residuos D -ala-D –ala; lo que conlleva a la presencia de cantidades reducidas de L-glutamina disponibles para la amidación del D-glutamato en el puente pentapéptido. Como resultado, hay más residuos D -ala-D -ala disponibles para enlazar y atrapar el antibiótico. A continuación, la vancomicina enlazada actúa como un impedimento para las nuevas moléculas de antibiótico que llegan a su destino en la membrana citoplasmática (8, 9).
La cromatografía líquida de alta presión (HPLC) proporciona una prueba más de este novedoso mecanismo de resistencia que muestra que grandes cantidades de vancomicina quedan atrapadas en el peptidoglucano anormal (3, 8, 9, 14). De hecho, se ha demostrado que se puede recuperar vancomicina intacta a partir de las paredes celulares de cepas VISA, lo que indica que el antibiótico no es desactivado, sino simplemente secuestrado por la bacteria. Además, las paredes celulares alteradas parecen tener una afinidad reducida para vancomicina, como objetivos solubles son capaces de enlazar más antibiótico en presencia de aislamientos de vancomina resistentes (7, 8).
En 1997, la primera cepa MU3, con susceptibilidad reducida a vancomicina y teicoplanina, fue reportada en Japón, seguida de MU50 (5). Las cepas VISA (intermedias a vancomicina) han sido reportadas a nivel mundial: USA (28); Francia (29); Corea del Sur (25); Reino Unido (30, 31); Suráfrica (32), Brasil (26), Grecia (33), Alemania (34) y China (35). Cepas de S. aureus con resistencia intermedia heterogénea (hVISA) han sido reportadas en varias partes del mundo (6).
La resistencia a vancomicina en S. aureus es difícil de definir principalmente debido a problemas metodológicos en su detección en el laboratorio clínico microbiológico (2, 8, 36). La prueba de difusión del disco, usando el disco estándar de 30 µg, frecuentemente falla al clasificar erróneamente aislamientos con susceptibilidad intermedia como completamente susceptibles, por lo que no se debe utilizar este método. Los métodos automatizados también tienen limitaciones en este sentido. Las cepas VISA no son confiablemente distinguidas de las sensibles por estos métodos. En la actualidad, la determinación de la concentración inhibitoria mínima, por dilución en caldo o agar, constituyen el gold standard para la determinación de susceptibilidad a vancomicina; sin embargo, estos métodos no están disponibles para uso rutinario en los laboratorios (37).
Hasta la fecha no existen métodos estandarizados para la detección de VISA o hVISA, por lo que se ha propuesto el perfil de análisis poblacional (PAP) como un método de screening. Recientemente, el criterio del área PAP bajo la curva (AUC) para la determinación de la resistencia a vancomicina es utilizado con radios PAP-AUC £ 0,90 para S. aureus sensible a vancomicina (VSSA); 0,90 – 1,30 para hVISA y ³ 1,3 para VISA (37).
Como se mencionó previamente, el mecanismo de resistencia en cepas VISA no está asociado con la existencia de ningún gen y ha sido objeto de investigación exhaustiva. Se han observado varias alteraciones fenotípicas en cepas VISA, que incluyen: colonias pequeñas, tasas de crecimiento menores, diámetros celulares mayores, engrosamiento de la pared celular, incremento en la síntesis de la pared celular, incremento en la incorporación de N-acetil-glucosamina y disminución de la sensibilidad a lisostafina (6).
Muchos reportes implican al gen mecA en el desarrollo de resistencia a vancomicina en cepas SAMR. Adhikari y cols. refieren que la adquisición de resistencia a vancomicina en cepas SAMR está relacionada con la deleción de mecA, sugiriendo que esta deleción acoplada con otros re-arreglos, puede ser responsable del incremento en el fenotipo de resistencia a vancomicina (38).
Análisis de hibridación genómica comparativa revelaron la supresión específica de un segmento de 1,8kb que abarca dos marcos abiertos de lectura adyacentes (ORFs) de función desconocida en un revertante teicoplanina-susceptible (Cepa 14-4rev) en comparación con la secuencia de su cepa parental isogénica, teicoplanina resistente (Cepa 14-4). Este hallazgo provocador requiere realizar un análisis genético detallado de la contribución de este segmento genómico a la resistencia a glicopéptidos. A pesar de reiterados esfuerzos en el laboratorio, 14-4 y 14-4rev se han mostrado refractarias a la mayoría de las manipulaciones genéticas. Para sortear esa dificultad, se evaluó la contribución de ambos ORFs (designados como factores trfA y trfB de resistencia a teicoplanina) sobre la resistencia a este antibiótico en un contexto diferente, genéticamente manejable. Análisis genéticos mostraron que mutaciones trfA y/o trfB simples o dobles abolieron la resistencia a teicoplanina en dos descendientes resistentes, independientes, derivados de la cepa NCTC8325 ISP794 generados por pasajes en medios con el antibiótico. La frecuencia de mutantes resistentes disminuyó marcadamente por la ausencia de trfAB en la cepa ISP794 teicoplanina-susceptible. Sin embargo, una baja tasa de mutantes resistentes a teicoplanina fue seleccionada de ISP794 trfAB, lo que indica una contribución adicional de factores adicionales independientes de trfAB en la aparición de la resistencia de bajo nivel a glicopéptidos. Aún más, se ha reportado que la mutación en trfAB podría afectar no sólo la resistencia a teicoplanina, sino también a vancomicina y oxacilina (4).
Nuevos glicopéptidos
Modificaciones en la estructura química de los glicopéptidos naturales han conducido al descubrimiento de una serie de compuestos con marcada actividad contra organismos grampositivos multirresistentes, incluyendo SAMR y EVR. Entre estos se encuentran los llamados lipoglicopéptidos: oritavancina, dalbavancina y telavancina (7).
Dalbavancina (Zeven®, Pfizer, Inc): La dalbavancina (B1 397) es un glicopéptido semi-sintético derivado de la teicoplanina; de la que difiere por la presencia de un ácido acil-amino-glucurónico en lugar de un sustituyente acil-glucosamina y, por una cadena lateral alifática (dimetil-aminopropilamina) unida al núcleo del péptido. Es más potente que los glicopéptidos originales (vancomicina y teicoplanina) contra los estafilococos; sin embargo, no es mejor que la teicoplanina frente a EVR. Hasta la fecha no se ha reportado resistencia a este fármaco (7).
Telavancina (Theravance, Inc): es un lipoglicopéptido estructuralmente similar a la vancomicina. Posee el mismo núcleo glicopéptido que la vancomicina; pero además, contiene una cadena lipofílica lateral (fosfonometil aminometil), la cual hipotéticamente, incrementa el anclaje del antibiótico en la membrana celular, favoreciendo la afinidad por el lípido II; mientras que la fracción polar, mejora las características farmacocinéticas de la droga (absorción, distribución, metabolismo y excreción renal) (7, 39).
Es un antibiótico rápidamente bactericida, su efecto es dependiente de la concentración y tiene una potente actividad contra patógenos grampositivos clínicamente importantes, tales como: estafilococos (incluyendo cepas SAMR, hVISA y VISA) y estreptococos. Su modo de acción es múltiple. En un mecanismo similar al de vancomicina; pero 10 veces más potente, inhibe la síntesis de la pared celular mediante uniones fuertes con la estructura central aglicona D-alanina-D-alanina del precursor, lípido II y los intermediarios nacientes del peptidoglucano; por lo que inhibe la etapa final de la biosíntesis de pared celular (transpeptidación y transglicosilación). Adicionalmente, en un mecanismo diferente no compartido con vancomicina, la telavancina interactúa con la membrana celular grampositiva para producir cambios en el potencial de membrana y alterar la permeabilidad celular. No se han reportado cepas resistentes a este antibiótico (7, 39).
Oritavancina (Targanta Therapeutics, Cambridge, MA, USA): se obtiene por alquilación reductiva de la cloro-oeremomicina, un producto natural de fermentación de Amycolaptosis orientalis, que difiere de la vancomicina por la sustitución de la vancosamina de la mitad disacárida por epivancosamina. Posee un espectro de acción similar a la vancomicina; pero con valores de CIM consistentemente inferiores a 1 mg/L. En contraste con la vancomicina, la oritavancina dimeriza fuertemente y es capaz de anclarse dentro de la membrana celular bacteriana. Se cree que las interacciones generadas, sean la base para su mayor actividad antibacteriana (7, 39, 40). Se ha indicado también que el disacárido sustituído r-cloro- fenil-bencil podría inhibir la síntesis de la pared celular a nivel de la transglicosilación; aún en ausencia de la unión de los precursores al depsipéptido (7).
En S. aureus, no se ha observado resistencia, incluso entre cepas VISA; sin embargo, in vitro, se han obtenido cepas de enterococos portadoras de los genes vanA y vanB con susceptibilidad disminuida a este antibiótico. Los mecanismos de resistencia incluyen: 1) bloqueo total de la entrada de precursores D-ala; 2) mutaciones en el sensor Van Sb del cluster Van B y 3) expresión de vanZ, cuya función precisa es desconocida (7, 39, 40).
Conclusión
La resistencia a la vancomicina en S. aureus está empezando a emerger como un problema clínico, pero la atención que ya ha recibido sirve para subrayar la seriedad del problema. Una mejor comprensión del mismo será clave para ayudar a prevenir y tratar estas infecciones. Si la experiencia pasada con microorganismos multirresistentes es un indicador, el problema de S. aureus vancomicina resistente sólo crecerá en el futuro. Sin embargo, una mayor conciencia y el cumplimiento estricto de las actuales directrices prácticas para el uso de la vancomicina y las prácticas del control de la infección pueden ayudar a limitar el impacto de estos organismos.
Referencias Bibliográficas
1. Bhateja P, Purnapatre K, Dube S, Fatma T, Rattan A. Characterization of laboratory-generated vancomycin intermediate resistant Staphylococcus aureus strains. Int J Antimicrob Agents 2006; 27(3):201-11. [ Links ]
2. Yusof A, Engelhardt A, Karlsson A, Bylund L, Vidh P, Mills K, et al. Evaluation of a New E-test Vancomycin-Teicoplanin Strip for Detection of Glycopeptide-Intermediate Staphylococcus aureus (GISA), in Particular, Heterogeneous GISA. J Clin Microbiol 2008; 46(9):3042-7. [ Links ]
3. Hiramatsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis 2001; (3):147-55. [ Links ]
4. Renzoni A, Kelley W, Barras C, Monod A, Huggler E, Francois P, et al. Identification by Genomic and Genetic Analysis of Two New Genes Playing a Key Role in Intermediate Glycopeptide Resistance in Staphylococcus aureus. Antimicrob Agents Chemother 2009; 53(3):903-11. [ Links ]
5. Hiramatsu K. Vancomycin resistance in staphylococci. Drug Resist Update 1998; 1(2):135-50. [ Links ]
6. Cui L, Lian J, Neoh H, Reyes E, Hiramatsu K. DNA Microarray-Based Identification of Genes Associated with Glycopeptide Resistance in Staphylococcus aureus. Antimicrob Agents Chemother 2005; 49(8):3404-13. [ Links ]
7. Finch R, Eliopoulos G. Safety and efficacy of glycopeptide antibiotics. J Antimicrob Chemother 2005; 55 Suppl 2:ii5-13. [ Links ]
8. Srinivasan A, Dick J, Perl TM. Vancomycin Resistance in Staphylococci. Clin Microbiol Rev 2002; 15(3):430-8. [ Links ]
9. Lowy F. Antimicrobial Resistance: the example of Staphylococcus aureus. J Clin Invest 2003; 111(9):1265-73. [ Links ]
10. Weigel L, Clewell D, Gill S, Clark N, McDougal L, Flannagan S, et al. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003; 302 (5650):1569-71. [ Links ]
11. Cetinkaya Y, Falk P, Mayhall C. Vancomycin - Resistant Enterococci. Clin Microbiol Rev 2000; 13(4):686-707. [ Links ]
12. Hiramatsu K, Cui L, Kuwahara-Arai K. Has vancomycin-resistant Staphylococcus aureus started going it alone? Lancet 2004; 364(9434):565-6. [ Links ]
13. Tenover F, Biddle J, Lancaster M. Increasing resistance to vancomycin and other glycopeptides in Staphylococcus aureus. Emerg Infect Dis 2001; 7(2):327-32. [ Links ]
14. Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover F. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 1997; 40(1):135-6. [ Links ]
15. Gemmell C. Glycopeptide resistance in Staphylococcus aureus: is it a real threat? J Infect Chemother 2004; 10(2):69-75. [ Links ]
16. Hiramatsu K, Okuma K, Ma X, Yamamoto M, Hori S, Kapi M. New trends in Staphylococcus aureus infections: glycopeptide resistance in hospital and methicillin resistance in the community. Curr Opin Infect Dis 2002; 15(4):407-13. [ Links ]
17. Hiramatsu K, Cui L, Kuroda M, Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol 2001; 9(10):486-93. [ Links ]
18. Foucault M, Courvalin P, Grillot-Courvalin C. Fitness Cost of VanA-Type Vancomycin Resistance in Methicillin-Resistant Staphylococcus aureus. Antimicrob Agents Chemother 2009; 53(6):2354-9. [ Links ]
19. Finan J, Archer G, Pucci M, Climo M. Role of Penicillin-Binding Protein 4 in Expression of Vancomycin Resistance among Clinical Isolates of Oxacillin-Resistant Staphylococcus aureus. Antimicrob Agents Chemother 2001; 45(11):3070-5. [ Links ]
20. Bischoff M, Roos M, Putnik J, Wada A, Glanzmann P, Giachino P, et al. Involvement of multiple genetic loci in Staphylococcus aureus teicoplanin resistance. FEMS Microbiol Lett 2001; 194(1):77-82. [ Links ]
21. Hanaki H, Kuwahara-Arai K, Boyle-Vavra S, Daum R, Labischinski H, Hiramatsu K. Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J Antimicrob Chemother 1998; 42(2):199-209. [ Links ]
22. Maki H, McCallum N, Bischoff M, Wada A, Berger-Bachi B. tcaA Inactivation Increases Glycopeptide Resistance in Staphylococcus aureus. Antimicrob Agents Chemother 2004; 48(6):1953-9. [ Links ]
23. Kuroda M, Kuwahara-Arai K, Hiramatsu K. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem Biophys Res Commun 2000; 269(2):485-90. [ Links ]
24. Cui L, Ma X, Sato K, Okuma K, Tenover F, Mamizuka E, et al. Cell Wall Thickening Is a Common Feature of Vancomycin Resistance in Staphylococcus aureus. J Clin Microbiol 2003; 41(1):5-14. [ Links ]
25. Kim M, Pai C, Woo J, Ryu J, Hiramatsu K. Vancomycin-Intermediate Staphylococcus aureus in Korea. J Clin Microbiol 2000; 38(10):3879-81. [ Links ]
26. Oliveira G, DellAquila A, Masiero R, Levy C, Gomes M, Cui L, et al. Isolation in Brazil of nosocomial Staphylococcus aureus with reduced susceptibility to vancomycin. Infect Control Hosp Epidemiol 2001; 22(7):443-8. [ Links ]
27. Cui L, Murakami H, Kuwahara-Arai K, Hanaki H, Hiramatsu K. Contribution of a Thickened Cell Wall and Its Glutamine Nonamidated Component to the Vancomycin Resistance Expressed by Staphylococcus aureus Mu50. Antimicrob Agents Chemother 2000; 44(9):2276-85. [ Links ]
28. Boyle-Vavra S, Labischinski H, Ebert CC, Ehlert K, Daum RS. A Spectrum of Changes Occurs in Peptidoglycan Composition of Glycopeptide-Intermediate Clinical Staphylococcus aureus Isolates. Antimicrob Agents Chemother 2001; 45(1):280-7. [ Links ]
29. Chesneau O, Morvan A, Solh N. Retrospective screening for heterogeneous vancomycin resistance in diverse Staphylococcus aureus clones disseminated in French hospitals. J Antimicrob Chemother 2000; 45(6): 887-90. [ Links ]
30. Hood J, Giles E, Cosgrove B, Curran E, Morrison D, Gemmell C. Vancomycine-intermediate Staphylococcus aureus at a scottish hospital. J Infect 2000; 40(2):A11. [ Links ]
31. Paton R, Snell T, Emmanuel F, Miles R. Glycopeptide resistance in an epidemic strain of methicillin-resistant Staphylococcus aureus. J Antimicrob Chemother 2001; 48(6): 941-2. [ Links ]
32. Watanakunakorn C. Mode of action and in-vitro activity of vancomycin. J Antimicrob Chemother 1984; 14 Suppl D:7-18. [ Links ]
33. Tsakris A, Papadimitriou E, Douboyas J, Stylianopoulou F, Manolis E. Emergence of vancomycin-intermediate Staphylococcus aureus and S. sciuri, Greece. Emerg Infect Dis 2002; 8(5):536-7. [ Links ]
34. Reipert A, Ehlert K, Kast T, Bierbaum G. Morphological and Genetic Differences in Two Isogenic Staphylococcus aureus Strains with Decreased Susceptibilities to Vancomycin. Antimicrob Agents Chemother 2003; 47(2):568-76. [ Links ]
35. Lu J, Lee S, Hwa S, Yang A. Septic Arthritis Caused by Vancomycin-Intermediate Staphylococcus aureus. J Clin Microbiol 2005; 43(8):4156-8. [ Links ]
36. Trakulsomboon S, Danchaivijitr S, Rongrungruang Y, Dhiraputra C, Susaemgrat W, Ito T, et al. First Report of Methicillin-Resistant Staphylococcus aureus with Reduced Susceptibility to Vancomycin in Thailand. J Clin Microbiol 2001; 39(2):591-5. [ Links ]
37. Walsh T, Bolmstrom A, Qwarnstrom A, Ho P, Wootton M, Howe R, et al. Evaluation of Current Methods for Detection of Staphylococci with Reduced Susceptibility to Glycopeptides. J Clin Microbiol 2001; 39(7): 2439-44. [ Links ]
38. Adhikari R, Scales G, Kobayashi K, Smith J, Berger-Bachi B, Cook G. Vancomycin-induced deletion of the methicillin resistance gene mecA in Staphylococcus aureus. J Antimicrob Chemother 2004; 54(2): 360-3. [ Links ]
39. Dunbar L, Tang D, Manausa R. A review of telavancin in the treatment of complicated skin and skin structure infections (cSSSI). Ther Clin Risk Manag 2008; 4(1):235-44. [ Links ]
40. Lentino J, Narita M, Yu V. New antimicrobial agents as therapy for resistant gram-positive cocci. Eur J Clin Microbiol Infect Dis 2008; 27(1):3-15. [ Links ]

