








Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Técnica de la Facultad de Ingeniería Universidad del Zulia
Print version ISSN 0254-0770
Rev. Téc. Ing. Univ. Zulia vol.28 no.2 Maracaibo Aug. 2005
Electrochemical studies of Fe(CN)64-/Fe(CN)63- on gold ultramicroelectrodes varying the concentrations of KF as supporting electrolyte
Sabino Menolasina
Departamento de Análisis y Control, Facultad de Farmacia, Universidad de los Andes, Mérida, Venezuela 501. E-mail: sabino@.ula.ve
Abstract
Gold disk ultramicroelectrodes of 10 mm diameter were fabricated using gold wires. These ultramicroelectrodes were characterized by electrochemical measurements and scanning electron microscopy (SEM). Electrochemical behavior of the hexacyano ferrate couple using gold ultramicroelectrodes (10 mm diameter) was investigated in a range of different concentrations of KF as supporting electrolyte. The steady state response at a gold ultramicrodisc at different concentrations of hexacyanoferrate couple with high concentrations of electrolyte was utilised to determine the diffusion coefficients of both electroactive species. Voltammetric experiments shown, that the oxidation and reduction of this couple is affected by the concentration of the supporting electrolyte. Theoretical Tafel plots were calculated considering double layer effects and comparisons with the experimental results indicated that determination of the heterogeneous rate constants for the free (unpaired) anions species seems to be imposible. Even at lower concentrations and after correction for double layer effects, the reaction is still dominated by ion pair effects.
Key word: Gold ultramicroelectrodes; scanning electron microscopy; Tafel plots.
Estudios electroquímicos del sistema Fe(CN)64-/Fe(CN)63- sobre ultramicroelectrodos de oro variando la concentración de KF como electrolito de soporte
Resumen
Ultramicroelectrodos de oro de 10 mm de diámetro fueron fabricados utilizando alambres de oro. Estos ultramicroelectrodos fueron caracterizados electroquimicamente y utilizando microscopía de barrido electrónico para determinar la forma real de la superficie y obtener información acerca de la calidad del sello entre la interface del metal y el material aislante utilidado en la construcción del electrodo. El comportamiento electroquímico de la pareja redox Fe(CN)64-/Fe(CN)63- fue investigado a diferentes concentraciones del electrolito de soporte KF. Los experimentos realizados por Voltamperometría mostraron que la oxidación y reducción de estas especies son afectadas por la concentración del electrolito de soporte. La velocidad de transferencia electrónica es favorecida al aumentar la concentración del electrolito de soporte. Cálculos teóricos de curvas Tafel tomando en cuenta los efectos de la doble capa fueron realizados y comparados con los resultados experimentales. Estos estudios mostraron que no se puede determinar las constantes de velocidad heterogénea para las especies iónicas no apareadas, debida a que aún a concentraciones muy bajas del electrolito de soporte, la reacción sigue siendo controlada por efectos de par iónico.
Palabras clave: Ultramicroelectrodos; microscopía de barrido electrónico; curvas Tafel.
Recibido el 13 de Septiembre 2004
En forma revisada el 13 de Junio 2005
Introduction
Electrochemical studies of the hexacyanoferrate couple in absence of supporting electrolyte KCl using carbon ultramicroelectrodes have shown that the reduction of Fe(CN)63- is suppressed completely [1]. This suppression was attributed to a “dynamic diffuse layer effect” in which the transport of charged reactants across the diffuse part of the double layer limits the rate of their electroreduction. However, other factors such as the kinetics of electron transfer at the surface could be involved. The influence of ion pair formation between the hexacyanoferrate couple and the alkali metal cations of the electrolyte on the rate and mechanism of the charge transfer has been established [2, 3]. First order dependence of the rate constant on cation concentration of the electrolyte was found by Peter et.al. [3] for this couple over a wide range of electrolyte concentrations. Other authors have suggested that the hexacyanoferrate electrode transfer process can also be governed by other factors besides the processes of charge transfer and diffusion [4, 5]. Adsorption of ferrocyanide and ferricyanide on the electrode surface has been reported. Fleischman et al. [4] studied this redox system in alkali chloride solutions using enhanced Raman spectroscopy (SERS) on gold surface. Beriet and Pletcher [5] have reported that the reduction of ferricyanide as well as the oxidation of ferrocyanide could be affected by surface poisoning on the platinum surface. However it is necessary to point out that the experiments carried out by Fleischmann et.al and Beriet and Pletcher were made using KCl as supporting electrolyte and it is known that chloride ions can be adsorbed at Pt and Au electrodes [6]. The adsortion of this anion can change the kinetic behaviour of the hexacyanoferrate redox system in solutions of low concentrations of supporting electrolyte. Although some authors have verified the presence of adsorbed species [4, 5] on Pt and Au electrodes, other authors have not found any evidence of specific adsorption [3] on gold electrodes when KF was used as supporting electrolyte. On the other hands, the hexacyanoferrate system is often considered as a ‘model’ redox system [7, 8].
All the research carried out on this system so far has shown that many problems related to the charge transfer mechanism still require investigation. In the present work the hexacyanoferrate couple was studied in a range of different concentrations of KF as supporting electrolyte. The aim was to obtain information that will allow an understanding of the relative importance of the effects produced when much, little or no supporting electrolyte was used, as well as to obtain more information about the charge transfer kinetics of this system.
Experimental
Reagent grade concentrated 70% HClO4, KF, K3Fe(CN)6, K4Fe(CN)6, were obtained from Fluka; All these chemicals were used as received. Supporting electrolyte solutions of KF were treated with purified active charcoal for gas adsorption (particle size 0.85-1.70 mm) from BDH to eliminate organic impurities. The fabrication of gold ultramicroelectrodes proved to be a difficult task. The main difficulty for reproducing a good seal stems from the relatively large difference between the thermal expansion coefficients of the gold and glass, which produces a large gap at the gold-glass interface. It was found that using a composite of AralditeTM CY1301 + Hardener HY 1300 from Ciba-Geigy Plastic, very good seals between the gold and the composite could be obtained. Gold discs were then fabricated using gold wires of 10 and 20 mm diameter, 99.99% (Goodfellow Metal). The gold ultramicroelectrodes were polished with different grades of alumina (Buehler) and using ultrapure water as lubricant.
The gold ultramicroelectrodes were treated electrochemically by cycling between 0 and 1.45 V vs. SCE in 0.2 mol dm-3 HClO4.
Electrochemical experiments were carried out using a single compartment cell with a two electrode configuration using a saturated calomel electrode (SCE) as reference electrode. Cyclic voltammetry experiments were carried out using a waveform generator (Hitek Instruments) coupled to a current amplifier with a low pass filter (sensitivity of 0.01mA V1), built in house. The amplifier and the cell were placed in an earthed metal Faraday box.
Results and Discussion
The electrochemical characterization was performed using HClO4 as supporting electrolyte because the range of potential where oxygen electrosoption occur can be reduced, hence increased surface roughening avoided. Figure 1 show the voltammogram obtained at a gold ultramicroelectrode in HClO4 aqueous solution. In the double layer region, an electrode process is observed around 0.65 V. This shoulder is attributed to the adsorption of the ClO4- anion [9]. In the oxide formation region, replacement of the adsorbed anions by OH- occurs and two peaks are observed. The first peak at 1.25 V is attributed to the formation of the first sublattice of OH- deposited in between adsorbed anions [9]:

[MxA-]M + H2O →[MxA-]MOH + H+ + e- (1)
where [MxA-]M represent the adsorbed anions on metal. The second peak at 1.35 V has been attributed to the deposition of OH- accompanied by desorption of the anions [10]:
MxA- + H2O Û Mx-1 + MOH + A- + H+ + e- (2)
A peak is observed on the cathodic side of the voltammogram between 0.85 and 0.9 V, which corresponds to the reduction of adsorbed oxygen. The shape of the voltammogram is similar to that reported for polycrystalline gold [11]. The effective area of the gold ultramicroelectrode was calculated taking into account that the cathodic charge in aqueous solutions of HClO4 increases almost linearly with the applied potential above 1.2 V and that this charge is independent of pH [12].
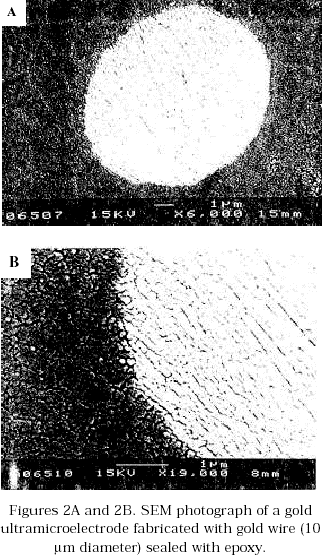
The charge value (QB) used was taken from the curve cathodic charge vs anodization potential reported by Brummer et.al. [12]. This value and the charge obtained experimentally by integration of the oxide stripping peak (Figure 3) were used to calculate the real area of the microdisc from the ratio: ![]() . For a series of experiments carried out at 60 mV/s, using gold wires of 10 mm diameter, the real area calculated was 77.38 ± 2.21 mm2. The quality of the seal between the electrode material and the insulating material as well as the shape of the microdisk were investigated by scanning electron microscopy (Figures 2A and 2B).
. For a series of experiments carried out at 60 mV/s, using gold wires of 10 mm diameter, the real area calculated was 77.38 ± 2.21 mm2. The quality of the seal between the electrode material and the insulating material as well as the shape of the microdisk were investigated by scanning electron microscopy (Figures 2A and 2B).

Steady state voltammetric studies to different concentrations of the hexacyanoferrate couple were performed keeping the concentrations of ferrocyanide equal to the concentration of ferricyanide to determine the diffusion coefficient of the oxidised and reduced forms (Figure 3a). In these experiments it was observed that the diffusion limited current was proportional to the concentration (Figure 3b). The diffusion coefficients of the oxidised and reduced species were determined from the slope of plots of diffusion current vs. concentration applying the equation:
![]()
The value obtained for the oxidised species was 5.74x10-6 ± 0.2x10-6 cm2 s-1 and the value for the reduced species was 7.25x10-6 ± 0.2x10-6 cm2 s-1. These values agree with the values reported in the literature [13].
Supporting electrolyte dependence
A very large number of experiments were carried out using different [electrolyte]/ [hexacyanoferrate couple] ratios. All the experiments were initiated at the equilibrium potential. In solutions where the concentration of supporting electrolyte was > 0.1 mol dm-3, well defined steady-state behaviour was observed using a gold ultramicroelectrode (Figure 4A). The equilibrium potential of the hexacyanoferrate couple shifted to more positive potential as the concentration of supporting electrolyte was increased beyond 0.1 mol.dm-3. Although the equilibrium potential had an unknown contribution from the liquid junction potential of the reference electrode (SCE), the changes observed seemed too large to be attributed to changes of liquid junction potential. The potential shift was attributed to the association of the K+ cations and the hexacyanoferrate ions. When the concentration of supporting electrolyte was < 0.1 mol dm-3 and the hexacyanoferrate couple concentration £ 1x10-3 mol dm-3, steady state limiting currents were not observed (Figure 4B).

According to Wightman et al. [14], electrochemical measurements performed when the electrolyte/analyte ratio is > 1 can give information which is not affected due to scavanging electrolyte in the diffuse layer. In a series of experiments the [electrolyte]/ [hexacyanoferrate couple] ratio was changed from 0 to 100, using a constant concentration of hexacyanoferrate. However no steady-state behaviour was observed. On the other hands, if migration effects are considered, the ratio ii /id (where ii is the limiting current when supporting electrolyte is absent from solution and id is the diffusion controlled limiting current in the presence of excess supporting electrolyte) must be > 1 for the anion oxidation, according the equations developed by Amatore et al. [15] for the oxidation or reduction of multiply charged ions in the absence of supporting electrolyte. For this reason comparisons with this model can not be made. Moreover, this model has been developed by making several assumptions such as similar diffusion coefficients for products and reactant, no double layer effects, and no ion pairing.
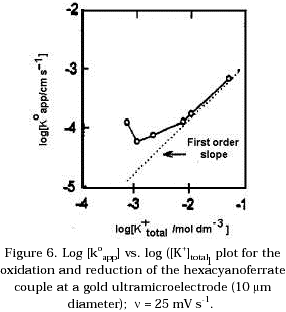
According to the results obtained by Campbell and Peter [16] for the system Fe(CN)64-/Fe(CN)63- on gold electrodes using impedance measurements and KF as supporting electrolyte, a minimum in the apparent rate constant is observed when the total concentration of K+ cations is increased over the range 4x10-4 to 1 mol dm-3. Experiments with different concentration of supporting electrolyte in the range where the total concentration of K+ increase from 7x10-4 to 0.01 mol dm-3 were carried out. Figure 5 shows Tafel plots under the experimental conditions mentioned above. From these plots, the apparent exchange current ioapp and the electron transfer coefficients were determined for both processes. It was found that the sum of the electron transfer coefficients for the anodic and cathodic processes is less than one. These results agree with the values obtained by Peter et al. [3] using a coulostatic method. Figure 6 shows log koapp vs. log [K+] total plots for the oxidation and reduction processes. The koapp was determined using the equation: ![]() . Where joapp is the exchange current density and C¥ is the concentration of the hexacyanoferrate couple in the bulk solution. A minimum in the apparent rate constant koapp is found when the total concentration of K+ is between 1x10-3 to 3x10-2 mol dm-3.
. Where joapp is the exchange current density and C¥ is the concentration of the hexacyanoferrate couple in the bulk solution. A minimum in the apparent rate constant koapp is found when the total concentration of K+ is between 1x10-3 to 3x10-2 mol dm-3.

Effects of association of the K+ cation with the hexacyanoferrate couple or double layer effects on the rate of electron transfer could be responsible for the behaviour observed in this range. According to Eaton et al. [17] the association constant K1 and K2 for the equilibrium reactions:

based on the extended Debye-Hückel theory; where K=K1 or K2, are the equilibrium constants depending on the equilibrium studied, and they are defined in terms of the molar concentration of the species present in the reaction; where N is a constant; N=4.08 for reaction (6), and N=3.06 for the reaction (7); I is the ionic strength. From this equation, values of K were determined for a constant concentration of the hexacyanoferrate couple and different concentrations of supporting electrolyte. From these values, the concentrations of KFe(CN)63- and KFe(CN)62- were determined.
Figure 7 shows the behavior of ion-pair association with the total concentration of K+ present in the system. Not surprisingly, it is observed that the ion association of the ferrocyanide anion occurs more appreciable than the association of the ferricyanide anion. When the total concentration of K+ is 2.2x10-3 mol dm-3, the extent of the association is 24% for the ferrocyanide anion, while it is only 4% for the ferricyanide anion (Figure 7).

From Figure 5 the Tafel plot for the electrochemical process in absence of supporting electrolyte shows the anodic and cathodic branches are not symmetrical. The anodic branch is higher than the cathodic branch. This behavior may be attributed to double layer effects.
For gold polycristalline surfaces the potential of zero charge is ~ -0.05 ± 0.1V vs. SCE [18]. This means the charge on the metal surface (qM) near the equilibrium potential is positive. This excess of positive charge on the metal will result in the concentration of ferrocyanide anions near to the electrode surface being higher than in a bulk solution and higher than the concentration of ferricyanide anions at the microelectrode surface. This difference in concentration produces that the current density at potentials positive of the equilibrium potential will be enhanced. The plots in Figures 8a and 8b were obtained using the following equation:
![]()
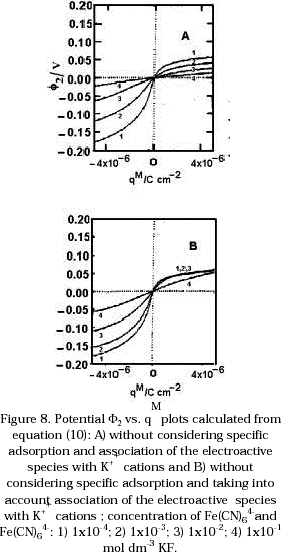
which is derived from the Gouy - Chapman theory of the diffuse double layer. The plots were calculated considering no association of the anion with the K+ cations (Figure 8a) and considering this association effects (Figure 8b). In this equation, ciS is the concentration of ions in bulk solution, and f2 is the potential in the outer-Helmholtz layer [19]. From these plots it is seen that when the electrode is charged positively, a high concentration of anions must be near to the electrode surface, which is reflected in the small values of ø2. If association of these electroactive species with the cations from the supporting electrolyte is considered, the concentration of these species near to the electrode surface is less with respect to the situation where no association is considered. This behaviour is also observed in the values of ø2 in Figure 8b.
The charge on the metal electrode can be related to the potential by the equation:
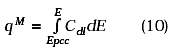
if Cdl is considered constant in the potential range studied, the potential diference E-Epzc is given by:
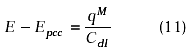
Figure 9A shows the behaviour of ø2 vs. E-Epzc calculated using this aproximation and Cdl equal to 25 mF cm-2.
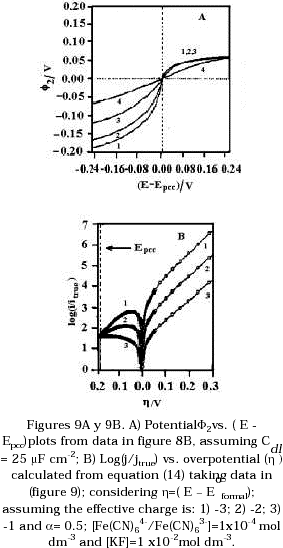
According to Figure 4A the equilibrium potential of the hexacyanoferrate couple shifted to more positive potential as the concentration of supporting electrolyte was increased. This mean that for building Tafel plots, the overpotential must be calculated taking into account the formal potential (Eo’), which is dependent on the medium since it includes the logarithmic activity coefficient terms (gi) as well as the standard electrode potential (Eo), according to the following relationship:
![]()
If the overpotential (h) is calculated as (E - Eo’) and this data and ø2 are taken from Figure 9A, the behavior of the ratio between the current density and the true exchange current density can be evaluated for the hexacyanoferrate couple at conditions where supporting electrolyte is present or absent using the equation:
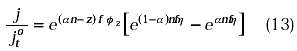
where jto is the true exchange current density, a is the cathodic transfer coefficient, z is the ionic valence of the oxidised species, f = F/RT, n is the number of electrons transfered. Figure 9B shows this behavior. It is seen that the theory predict that the cathodic current should be considerably hindered by double layer effects even taking into account the formation of ion-pair. Experimentally this effect was not as large as predicted in Figure 9B. The ion - pair species may be reacting faster than the free ferricyanide anions present near to the electrode- solution interface. The association constant of formation of ion - pair species may be different in the double layer from the association constant of these species in the bulk solution. The concentration of the ion - pairs species in equilibrium with the free association species may be affected in the double layer region due to the potential difference developed in the electrode - solution interface.
Electrochemical and chemical reactions could be also occurring in parallel during the oxidation and reduction of the hexacyanoferrate couple, influencing the electrochemical response observed experimentally.
Conclusión
Microelectrode studies of the oxidation and reduction of hexacyanoferrate couple have shown that the electrochemical process is affected by the concentration of KF the supporting electrolyte. The experimental results show that a low concentration of potassium ion and overall ionic strength, the electrode kinetics become dominated by double layer effects. At higher concentration of potassium ion, the apparent rate constant increases approximately linearly with [K+] as has been reported previosly [6]. Attempts to model the double layer effects on assuming that the reaction involves the free hexacyanoferrate ions showed that the predicted influence of the potential was larger than that observed experimentally. This provides evidence that the main reacting species are ion pairs, even at these low ionic strengths. The reason for this apparent anomaly is that the rate constants for the ion pair species are considerably higher than those for the free ions. Therefore the electrode reaction proceeds via the ion pairs, even when they are minority species. The experimental Tafel plots gave a and (a -1) values that do not sum to unity. This effect has been reported previously for measurements made by the coulostatic metods at much higher concentrations of KF [6]. The reason for this anomalous behaviour remains obscure. It is certainly not due to a preceding rate limiting chemical step involving ion pair formation, since this is expected to occur with a second order rate constant in excess of the diffusion controlled limit of 1010 dm3 mol s-1. Tafel plots predicted for taking into account the double layer effect show a large effect only on the cathodic branch. By contrast the anodic branch is largely unaffected because f2 is small. It therefore seems unlikely that double layer effects are responsables for the observed values of a. The hexacyanoferrate system is often considered as a ‘model’ redox system. However this study has revealed that the system is extremely complex. Comparison of experimental rate constant with values predicted by Marcus theory [20] are clearly not justified because the reacting species are ion pairs and the apparent rate constant is sensitive to the total potassium ion concentration.
In conclusion, this study has shown that it appears impossible to determine the rate constants for the hexacyanoferrate couple for the free (unpaired) anionic species. Even at the lower concentrations and after correction for double layer effects, the reaction is still dominated by ion pair effects.
Acknowledgements
I would like to acknowledge the Universidad de Los Andes and the Concejo Nacional de Investigaciones Científicas y Tècnologicas (CONICIT) for their financial support and is particularly grateful to Professor Laurence Peter, Bath University, for his valuable advice and for helpful discussions.
References
1. Lee C., Anson F.C., “Inhibition of the electroreduction of Fe(CN)63- at microelectrodes in the absence of supporting electrolyte-mediation of the inhibited reduction by methyl viologen”. J. Electroanal. Chem., Vol. 323, (1992), 381-389. [ Links ]
2. Bieman D.J., Fancett W.R., “The influence of ion pairing in the mechanism of electroreduction of anions”. J Electroanal.Chem., Vol. 34, (1972), 27-34. [ Links ]
3. Peter L.M., Durr W., Bindra P., Gerischer H., “Influence of alkali-metal cations on rate of Fe(CN)63-/Fe(CN)64-electrode process”. J.Electroanal., Chem., Vol. 71, (1976) 31-50. [ Links ]
4. Fleischmamn M, Graves P.R., Robinson J., “The Raman-Spectroscopy of the ferricyanide ferrocyanide system at gold beta-palladium hydride and platinum-elecrtrodes” J. Electroanal. Chem., Vol 182, (1985), 87-98. [ Links ]
5. Beriet C., Pletcher D., “A microelectrode study of the mechanism and kinetics of the ferro-ferricyanide couple in aqueous-media-the influence of the electrolyte and its concentration”. J. Electroanal. Chem., Vol. 361, (1993), 93-101. [ Links ]
6. Shi Z., Wu S., Lipkowki J., “Investigations of Cl- adsorption at the Au(111) electrode in the presence of underpotentially deposited copper atoms”. J. Electroanal. Chem., Vol. 384, ( 1995) 171-177. [ Links ]
7. Ertl P.; Robello E., Battaglini F., Mikkelsen S., “Rapid Antibiotic Susceptibility Testing via Electrochemical Measurement of Ferricyanide Reduction by Escherichia coli and Clostridium sporogenes”. Analytical Chem., Vol. 72, (2000),4957-4961. [ Links ]
8. Rosseinsky D., Glasser L., Brooke H. D., “Thermodynamic clarification of the curious ferric/potassium ion exchange accompanying the electrochromic redox reactions of Prussian Blue, iron(III) hexacyanoferrate(II)”. J. of the American Chemical Soc., Vol 126, (2004) 10472-10477. [ Links ]
9. Kozlowska H.A., Conway B.E., “Elementary steps of electrochemical oxidation of single-crystal planes of Au-I. Chemical basis of processes involving geometry of anions and the electrode surfaces”. Electrochim. Acta, Vol. 31, (1986) 1051-1061. [ Links ]
10. Laitinen H.Q.A., Chao S. M.; “The anodic surface oxidation of gold”. J. Electrochem. Soc., Vol. 108, (1961) 726-731. [ Links ]
11. Hoare J. P., “A cyclic voltammetric study of the gold-oxygen system”. J. Electrochem. Soc., Vol. 131, (1984) 1808-1815. [ Links ]
12. Brummer S.B., Makrides A.C., “Surface oxidation of gold electrodes”. J. Electrochem. Soc., Vol. 111, (1964) 1122-1128. [ Links ]
13. Kunimatsu K., Shigematsu Y., Uosaki K., Kita H., “Study of the Fe(CN)63-/Fe(CN)64- redox system on Pt by EMIRS.1. Infrared-Spectra of the intermediates in the charge-transfer”. J. Electroanal. Chem., Vol. 262, (1989) 195-209. [ Links ]
14. Drew S.M., Wightman R.M., Amatore C.A., “Voltammetry of ferrocene in low electrolyte-solutions”. J. Electroanal. Chem., Vol. 317, (1991) 117-124. [ Links ]
15. Amatore C., Fooset B., Bartelt J., Deakin M.R., Wightman R.M., “Electrochemical kinetics at microelectrodes.5. Migrational effects on steady or quasi-steady-state voltammograms”. J. Electroanal. Chem., Vol. 256, (1988), 255-268. [ Links ]
16. Campbell S.A., Peter L.M., “The effect of [K+] on the heterogeneous rate-constant for the Fe(CN)63-/Fe(CN)64- redox couple investigated by AC-Impedance Spectroscopy”. J. Electroanal. Chem., Vol. 364, (1994) 257-260. [ Links ]
17. Hanania G.I.H., Irvine D.H., Eaton W.A., George P., “Thermodynamic aspects of potassium hexacyanoferrate(3)-(2) system.2. Reduction potential”. J. Phys. Chem., Vol. 71, (1967) 2016-2022. [ Links ]
18. Clavillier J., Vanhuong N., “Study of the polycrystalline gold interface in contact with aqueous solutions of potassium perchlorate and perchloric acid”. J. Electroanal. Chem.Interfacial Electrochem., Vol. 80, (1977) 101-114. [ Links ]
19. Delahay P., Double layer and Electrode Kinetics, Interscience, New York, 1965, Ch.3,4,7,8,9. [ Links ]
20. Hale J.M., Reactions of molecules at electrodes, N.S. Hush, Ed.; Wiley-Interscience, 1971, pp 229. [ Links ]