










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInvestigación Clínica
versión impresa ISSN 0535-5133versión On-line ISSN 2477-9393
Invest. clín v.51 n.2 Maracaibo jun. 2010
Cincuenta años de uso clínico de la warfarina.
Jesús Alberto Quintero-González.
Instituto de Investigaciones Clínicas “Dr. Américo Negrette”, Facultad de Medicina, Universidad del Zulia. Maracaibo, Venezuela.
Autor de correspondencia: Jesús Quintero. Instituto de Investigaciones Clínicas “Dr. Américo Negrette”, Facultad de Medicina, Universidad del Zulia. Maracaibo, Venezuela. Correo electrónico: jequin@gmail.com.
Resumen. La warfarina es el anticoagulante oral (AO) más utilizado en la profilaxis a largo plazo de las complicaciones tromboembólicas que acompañan a diversas enfermedades. Sus indicaciones se han ampliado en los últimos años, a medida que son detectadas nuevas situaciones clínicas que predisponen a sufrir eventos trombóticos. Debido a sus características especiales, tales como: dosis muy variable en cada individuo, estrecho margen entre la dosis adecuada y la inadecuada, interacciones con múltiples fármacos, interferencia de su efecto por el alto consumo de vitamina K en la dieta y la posibilidad de que aparezcan complicaciones hemorrágicas o recurrencia de la trombosis, el empleo de este medicamento requiere un estricto control de su dosificación y una continua vigilancia, tanto desde el punto de vista clínico como de laboratorio. A pesar de sus numerosas desventajas y de tener más de cincuenta años de uso clínico, la warfarina no ha sido sustituida hasta la fecha, por los anticoagulantes orales de reciente aparición. En el año 1999, la warfarina llegó a ocupar el décimo primer lugar entre los medicamentos de mayor consumo en el mundo.
Palabras clave: warfarina, anticoagulantes orales, trombosis, INR.
Fifty years of clinical use of warfarin.
Abstract. Warfarin is the most utilized oral anticoagulant for the long term prophylaxes of thrombosis. Its use has been increased as new clinical conditions, capable of leading to thrombosis, have been detected. Due to the special characteristics of warfarin, such as the variability of doses for each individual, the narrow margin between adequate and inadequate doses, interaction with multiple pharmaceutical products, interference of its action by vitamin K present in the diet and the possibility of hemorrhagic complications or thrombotic recurrence, this drug requires a very careful dosage and strict laboratory and clinical monitoring. Despite being in the market for more than de fifty years and its many disadvantages, warfarin has not been substituted for the new oral anticoagulants. In 1999, warfarin was positioned eleventh on the list of the most used medicines in the world.
Key words: Warfarin, oral anticoagulant, thrombosis, INR.
RESEÑA HISTÓRICA
En la segunda década del siglo pasado, los pobladores de las llanuras de Canadá y del norte de los Estados Unidos comenzaron a sembrar trébol dulce, puesto que esta planta crecía muy bien en suelos pobres. Inmediatamente, el ganado comenzó a morir por hemorragias incontrolables debidas a lesiones leves o hemorragias internas sin signos externos de agresión. En 1922, Schofield (1), observó que el ganado estaba consumiendo una mezcla de trébol dulce que funcionaba como un potente anticoagulante y por esta razón, esta patología hemorrágica fue denominada “enfermedad del trébol dulce”. En 1931, Roderick (2), logró demostrar que los animales afectados por esta enfermedad presentaban disminución de la concentración plasmática de protrombina. No fue sino hasta 1941, cuando Link, Campbell y un grupo de químicos investigadores de la Universidad de Wisconsin (3, 4), aislaron y caracterizaron el agente hemorrágico contenido en el trébol dulce. Estos investigadores establecieron que este anticoagulante era el 3,3’-metilenobis-(4-hidroxicumarina), que más tarde se conocería como dicoumarol o bishidroxicumarina, y que sería el primer compuesto de esta familia de sustancias en ser comercializado. Link y su equipo, continuaron desarrollando anticoagulantes sintéticos más potentes basados en la estructura molecular del dicoumarol, con la finalidad de utilizarlos como venenos contra roedores, hasta que obtuvieron en 1948, la warfarina. El nombre de warfarina proviene del acrónimo WARF, de Wisconsin Alumni Research Foundation, más la terminación arina, que indica su relación con la cumarina. La warfarina se registró para uso comercial como raticida por primera vez en los Estados Unidos en 1948 y debido a que en 1951 un soldado norteamericano intentara suicidarse sin éxito con esta droga, comenzaron los estudios para establecer su utilidad como anticoagulante terapéutico. En 1954 fue aprobado su uso clínico en humanos (5). Sin embargo, el mecanismo de acción de este fármaco se desconoció hasta 1978, cuando se demostró su acción inhibitoria sobre el metabolismo de la vitamina K (VK) en el hígado.
ASPECTOS QUÍMICOS Y FARMACOCINÉTICOS DE LA WARFARINA
La warfarina sódica o 3-(a-acetonilbencil)-4-hidroxicumarina, es una mezcla racémica de dos isómeros ópticamente activos: las formas R y S. Estos enantiómeros difieren en su potencia anticoagulante, metabolismo, eliminación e interacciones con otros fármacos (6). Aunque su disponibilidad es completa cuando se administra por vía intravenosa, intramuscular, oral o rectal, sólo la forma oral está disponible para uso clínico. La absorción gastrointestinal es completa, aunque el pico de concentración plasmática suele alcanzarse entre 2 y 8 horas luego de una sola dosis oral. En el plasma fetal las concentraciones se aproximan a los valores maternos (7). La vida media plasmática (T½) es muy variable y oscila entre 36 y 42 horas, lo que le proporciona una gran ventaja para el control de su efecto anticoagulante, al compararse con otros derivados cumarínicos cuyas T½ son muy cortas o demasiado largas. La alta tasa de unión a la albúmina plasmática (99%), es la responsable de su larga T½ y que además, difunda muy poco al líquido cefalorraquídeo, orina y leche materna (8), por lo que puede ser usada sin riesgo durante la lactancia.
La forma S es tres veces más activa que la R, pero es eliminada más rápidamente (9). Este isómero es metabolizado por las enzimas microsomales hepáticas (citromo P450) a metabolitos hidroxilados inactivos (hidroxicumarinas) que son excretados en la bilis. En cambio, la forma R es metabolizada por enzimas solubles citosólicas y convertida a alcoholes de warfarina, los cuales son excretados en la orina y poseen mínima actividad anticoagulante (10). Entre las isoenzimas del citocromo P450 involucradas en el metabolismo de la warfarina se incluyen: 2C9, 2C19, 2C8, 2C18, 1A2 y 3A4. La 2C9 probablemente es la principal isoenzima que modula su actividad anticoagulante in vivo. El estudio del metabolismo de los diferentes isómeros alcanza gran importancia cuando se consideran las interacciones con otras drogas.
INTERACCIONES DE LA WARFARINA CON OTRAS DROGAS
La capacidad de interactuar con otros medicamentos, constituye una de las principales desventajas del uso de warfarina. Esta situación suele tener mayor relevancia en los pacientes que requieren terapia a largo plazo, en individuos de edad avanzada o en aquellos que presentan otras patologías que ameriten algún tipo de tratamiento médico. Es indudable que en los sujetos que consumen varios fármacos, la posibilidad de mantener un adecuado control del efecto anticoagulante es baja. Koch-Weser (11), observó que la interacción medicamentosa es la responsable del 25% de las complicaciones hemorrágicas en pacientes hospitalizados. Otros autores han descrito que el 33% de los medicamentos indicados por un médico interactúa con la warfarina y en el caso de los que se usan sin prescripción médica es del 30% (12).
Muchos han sido los mecanismos propuestos para explicar esta interacción. Entre ellos se incluyen: Interferencia en la absorción gastrointestinal, en la unión a proteínas plasmáticas, en el metabolismo y en su excreción. Las drogas que interfieren con el metabolismo de la warfarina pueden incrementar las concentraciones plasmáticas de ésta y en consecuencia, potenciar el efecto anticoagulante. Si se trata de medicamentos que afectan el isómero S, estos podrían tener mayor repercusión sobre el tiempo de protrombina (TP) y su efecto anticoagulante hacerse más evidente clínicamente en forma de hemorragias. Los fármacos que inducen activación de las enzimas microsomales hepáticas y los que interfieren con su absorción, pueden disminuir las concentraciones plasmáticas de warfarina, y por lo tanto, su efecto anticoagulante.
Desafortunadamente, la lista de fármacos que interactúan con la warfarina es extensa y aumenta continuamente con el advenimiento de nuevos productos. Las interacciones medicamentosas más importantes de este anticoagulante con sus respectivos mecanismos de acción se agrupan en la Tabla I.
INTERACCIONES MEDICAMENTOSAS DE LA WARFARINA
| Droga | Mecanismo | Referencia |
| Antibióticos Cefalosporinas (2 y 3 generación) Rifampicina Metronidazol Cloranfenicol Eritromicina Sulfas (clotrimoxazol) Azitromicina | Inhibición enzimática del ciclo de la VK Inducción de enzimas microsomales hepáticas Inhibición del isómero S Desconocido Inhibición de las enzimas microsomales hepát. Desplazamiento de unión a proteínas Inhibición de enzimas microsomales hepáticas | Beshtold (13) O’Really (8) O’Really (14) Christenson (15) Weibert (16) O’Really (17) Woldvredt (18) |
| Antiinflamatorios no esteroideos Acido acetilsalicílico Acido mefenámico Celecoxib Diflunisal Lornoxicam Piroxicam Rofecoxib Sulindac | Desplazamiento de unión a proteínas Otros efectos hemostáticos | Fausa (19) Seller (20) Mersfelder (21) Tempero (22) Ravic (23) Rhodes (24) Schwartz (25) Carter (26) |
| Antiarrítmicos Amiodarona Quinidina | Desplazamiento de unión a proteínas Inhibición del citocromo P450 CYP2 Desplazamiento de unión a proteínas | Rees (27) Gazzaniga (28) |
| Anticonvulsivantes Carbamacepina Fenitoína | Inducción del metabolismo Desconocido | Hansen (29) Nappi (30) |
| Antifúngicos Fluconazol Griseofulvina Miconazol | Inhibición de enzimas microsomales hepáticas Inducción de enzimas microsomales hepáticas Inhibición de enzimas microsomales hepáticas | Black (31) Cullen (32) O’Really (33) |
| Antihipertensivos Propanolol-Metoprolol Espironolactona | Disminución del metabolismo hepático Aumento de la síntesis de factores de coagul. | Scott (34) O’Really (35) |
| Antiulcerosos Cimetidina Omeprazol Sucralfato | Inhibición de la hidroxilación del isómero S Inhibición del metabolismo Probable inhibición de la absorción | Serlin (36) Sutfin (37) Braverman (38) |
| Antineoplásicos Tamoxifeno Interferon | Desconocido Inhibición enzimática | Lodwick (39) Adachi (40) |
| Hipolipemiantes Clofibrato Colestiramina Fluvastatina Sinvastatina - Lovastatina | Desconocido Inhibición de la absorción Inhibición del citocromo P450 CYP2C9 Desconocido | Oliver (41) Gross (42) Trilli (43) Gaw (44) Ahmad (45) |
| Alopurinol | Inhibición de metabolismo | Rawlins (46) |
| Hormonas Esteroides anabólicos Glucagón Tiroxina Aminoglutetimida | Desconocido Aumento de la afinidad por el receptor Aumento del catabol. de factores II, VII, IX y X Inducción de enzimas microsomales hepáticas | Acomb (47) Koch-Weser (48) Owens (49) Bruning (50) |
| Barbitúricos | Inducción de enzimas microsomales hepáticas | Lewis (51) |
| Acarbosa | Desconocido | Morreale (52) |
| Disulfiram | Inhibición del metabolismo | Rothstein (53) |
| Sulfinpirazona | Desplazamiento de unión a proteínas Inhibición de metabolismo | Davis (54) |
MECANISMO DE ACCIÓN DE LA WARFARINA
Los derivados cumarínicos son antagonistas de la VK, debido a que actúan sobre los factores de coagulación que dependen de esta vitamina para su activación. Algunos de estos factores poseen actividad procoagulante (II, VII, IX y X) y otros por el contario, funcionan como anticoagulantes naturales (proteína C, proteína S y proteína Z). Para que estas proteínas se vuelvan fisiológicamente activas, se requiere un cambio conformacional en su estructura, que consiste en la carboxilación de los residuos de ácido glutámico de la cadena N terminal, formándose así el ácido g-carboxiglutámico (Gla). Estos Gla le dan a la molécula la capacidad de ligarse a cationes bivalentes como el calcio y a través de éstos, a los fosfolípidos de membranas celulares, superficies donde se ensamblan los complejos macromoleculares de la coagulación (55).
Este proceso de carboxilación posribosomal, es dependiente del ciclo de la VK, el cual tiene lugar en el hígado. Una carboxilasa (g-glutamil-carboxilasa), es la encargada de llevar a cabo esta reacción y precisa de VK como cofactor, además de oxígeno molecular y dióxido de carbono. En presencia de la forma activa de esta vitamina (KH2 o hidroquinona), la reacción enzimática genera g-carboxiglutamato (Gla) y 2,3 epóxido de VK. Este último, es reciclado a VKH2 por acción de dos enzimas en forma sucesiva. La primera, la epóxido reductasa (VKOR) reduce el 2,3 epóxido de VK a VK1 (forma inactiva), mientras que la segunda, la quinona reductasa (VKQR) reduce la VK1 a VKH2 (56). La warfarina inhibe a la VKOR y en menor grado, a la VKQR, bloqueando este ciclo y por lo tanto, no se produce la reducción de la VK a su forma activa (57). No obstante, aún en ausencia de VKH2, estos factores se siguen sintetizando y aunque desde el punto de vista inmunológico son idénticos a los normales, carecen de su función procoagulante o anticoagulante. Estas proteínas que circulan acarboxiladas se conocen con el nombre de PIVKAS (proteins induced by vitamin k absence) (58).
En conclusión, el mecanismo de acción de la warfarina reside en su capacidad de interferir con el ciclo de conversión de la VK, lo que impide la g-carboxilación de los factores de coagulación, dando como resultado la producción hepática de factores acarboxilados o parcialmente carboxilados con actividad coagulante reducida. En la Fig. 1, se esquematiza el ciclo de la VK y el sitio de acción de los derivados cumarínicos.
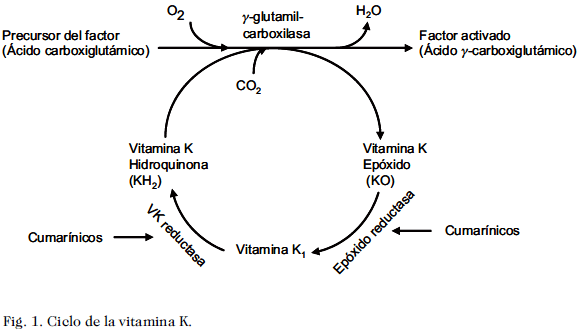
El efecto anticoagulante de los derivados cumarínicos depende de la depuración de los factores carboxilados circulantes, que a su vez depende de la T½ de cada uno de ellos. Esto explica claramente, la demora existente entre el momento en que se logra la máxima concentración plasmática del medicamento y su efecto anticoagulante máximo.
EFECTO ANTICOAGULANTE DE LA WARFARINA
Luego de la administración de la warfarina la actividad de los factores VK dependientes, disminuye de acuerdo a su T½, pudiendo variar desde 6 horas aproximadamente para el factor VII hasta 60 horas para el II. Esto significa, que se requiere de 3 a 5 días para que todos estos factores, alcancen su nivel más bajo de actividad después de iniciada la terapia con anticoagulantes orales (TAO). Como el factor VII tiene la T½ más corta, su inhibición es la responsable de la prolongación temprana del TP que se puede observar incluso en las primeras 24 horas y que podría ser interpretada erróneamente como una adecuada anticoagulación. Sin embargo, el efecto anticoagulante óptimo se alcanza cuando el TP se encuentra dentro de un rango terapéutico específico por varios días (59) y esto suele ocurrir después de las 72 horas, cuando ya se han inhibido todos los factores VK dependientes.
La proteína C (PC), uno de los principales inhibidores naturales de la coagulación, posee al igual que el factor VII una T½ muy corta. Este hecho, de gran importancia por sus implicaciones clínicas, podría contrarrestar el efecto anticoagulante ocasionado por la disminución del factor VII en las primeras horas y crear por el contrario, un estado protrombótico en pacientes con deficiencia de esta proteína. Es por ello, que estos individuos pueden desarrollar necrosis cutáneas en lugar de hemorragias cuando reciben TAO, especialmente cuando las dosis de inicio son altas (60).
PRUEBAS DE LABORATORIO UTILIZADAS PARA MEDIR EL EFECTO ANTICOAGULANTE
Debido a la capacidad que tiene la warfarina de prolongar el TP, esta prueba fue usada durante muchos años para el seguimiento del tratamiento anticoagulante oral. No obstante, los resultados expresados a través de este método, originaban una serie de inconvenientes debido a que podían variar de un laboratorio a otro, dependiendo del origen y calidad de la tromboplastina utilizada para su determinación. Las tromboplastinas pueden aislarse de diferentes especies (conejo, bovino y humana) y además, se emplean distintos métodos para su obtención. Por ejemplo, las tromboplastinas de conejo son menos sensibles al efecto anticoagulante de la warfarina que las provenientes de humanos y por otra parte, los tejidos de donde fueron extraídas (cerebro, pulmón y placenta), también les confieren propiedades particulares. Es decir, que para un mismo nivel de anticoagulación, una tromboplastina poco sensible podría prolongar poco el TP, mientras que otra de mayor sensibilidad lo haría de manera más pronunciada. Estas observaciones permitieron demostrar que la sola prolongación del TP, no es un buen parámetro para el seguimiento de los pacientes que reciben TAO y actualmente no se usa para este fin.
Muchos intentos se realizaron para tratar de resolver el problema de la variabilidad en la respuesta debida a las tromboplastinas, hasta que en 1983 el Comité de Referencia de la Comunidad Económica Europea en colaboración con el Comité Internacional para Estandarización en Hematología y la OMS (61), establecieron un modelo basado en la regresión lineal de los logaritmos del TP del paciente/TP de control (P/C) conocida como razón o R. Por otra parte, se crearon preparaciones internacionales de tromboplastina de referencia para la comparación según la especie de la cual provenga ésta. Al proyectar en un plano cartesiano, el logaritmo del TP de la tromboplastina de referencia internacional en el eje de las ordenadas y el logaritmo del TP de la tromboplastina de trabajo en el eje de las abscisas, la sensibilidad de esta última a la inhibición de los factores VK dependientes, viene dada por la pendiente de la línea de regresión a la que se le llamó Índice de Sensibilidad Internacional (ISI). Elevando la razón P/C al valor de ISI hallado, se calcula el valor que se obtendría de realizar la determinación del TP con la preparación internacional de referencia, y a este valor se le denominó INR (del inglés: International Normalized Ratio). Esta forma particular de expresar los resultados del TP es la correcta cuando se trata de controlar la TAO (62). La fórmula para obtener el INR se puede resumir de la siguiente manera:
INR = (TP paciente / TP control) ISI
Las preparaciones comerciales de tromboplastinas tienen establecido su valor de ISI, de acuerdo a la tromboplastina de referencia. El ISI más sensible es de 1, que corresponde a la trompoblastina humana de referencia internacional (OMS, 67/40). Se recomienda que los laboratorios trabajen con un ISI menor o igual a 1,2, tomando en cuenta que entre más cercano a 1 sea éste, la calidad del reactivo será mejor. Actualmente, la mayoría de los laboratorios, unidades y centros especializados en el control de TAO utilizan tromboplastinas recombinantes para la determinación del INR, que tienen un ISI muy cercano a 1.
La conversión del TP a INR pierde parcialmente confiabilidad con algunos tipos de sistemas automatizados o cuando la tromboplastina es muy poco sensible (ISI alto). Es recomendable usar un valor de TP obtenido de una mezcla de plasmas procedentes de voluntarios sanos como tiempo control o testigo, que debe ser determinado en cada laboratorio, y para cada lote de reactivo de tromboplastina que se utilizará.
Los valores de INR que indican el margen terapéutico adecuado para cada afección trombótica, han sufrido modificaciones desde que se introdujo esta modalidad, pero en la actualidad, están claramente establecidos.
INICIO Y CONTROL DE LA TAO
Inmediatamente que se presenta la trombosis venosa, debe instaurarse la terapia con warfarina. La misma se inicia con una dosis baja, que es suficiente para lograr la anticoagulación en la mayoría de los pacientes. No deben emplearse dosis de carga, ya que pueden desencadenar hemorragias o necrosis cutáneas (60). O’Reilly y Aggeler (63) demostraron que una dosis inicial alta no produce una inhibición más rápida de los factores II, VII, IX y X al compararse con una dosis baja. Se debe comenzar con 5 a 10 mg de warfarina, considerando que en individuos de edad avanzada, con bajo peso, con hepatopatías o en otras situaciones clínicas y en niños, pudiera ser necesario iniciar con dosis menores por el riesgo de aparición de hemorragias. La Sociedad Británica de Hematología recomienda iniciar la terapia con 10 mg durante dos días y ajustar la dosis al tercer día según el valor del INR (64). Sin embargo, la dosis usual de inicio empleada en la mayoría de las situaciones clínicas es de 5 mg.
Indudablemente, la experiencia del médico juega un papel preponderante a la hora de elegir la dosis inicial, ya que ésta es muy flexible dada la gran variabilidad en la respuesta. En otras palabras, la dosis debe ser individualizada con el fin de proveer una rápida pero segura anticoagulación, evitando las complicaciones y secuelas de la trombosis y disminuyendo el riesgo de sangramiento asociado a la terapia anticoagulante.
El inicio de la TAO debe ser simultáneo con heparina, y una vez alcanzados los límites terapéuticos de INR, ello significa tener 2 días continuos un INR entre 2 y 3 o lo que es más adecuado (aunque no rutinario), que el nivel de los factores VK dependientes se encuentre por debajo de 30% (lo que puede llevar alrededor de 5 días), se suspende la terapia con heparina. El primer control de INR deberá solicitarse entre el 3er y 4to día, ya que el nivel terapéutico se logra después de 72 horas (65). En los pacientes hospitalizados los controles sucesivos pueden realizarse cada 3 ó 4 días, mientras que los ambulatorios pueden ser controlados semanalmente o cada 2, 4 ó 6 semanas como máximo, lo que dependerá lógicamente de alcanzar el nivel deseado de INR, de acuerdo a la patología tratada. Las modificaciones en las dosis de aquellos pacientes que se encuentren fuera del rango terapéutico deberán ser evaluadas luego del 4to día. El uso concomitante de heparina, cuando se emplea la warfarina con fines profilácticos no es necesario, especialmente en las situaciones donde la duración de este tratamiento sea prolongada.
El rango de INR actualmente aceptado para la mayoría de las situaciones clínicas que ameritan anticoagulación de manera terapéutica y profiláctica, se ubica entre 2 y 3 (64).
INDICACIONES CLÍNICAS DE LA WARFARINA
La warfarina es el anticoagulante oral de elección en la prevención primaria y secundaria a corto y largo plazo de eventos trombóticos en pacientes con tromboembolismo venoso, fibrilación auricular, válvulas cardíacas protésicas, enfermedad arterial coronaria y en individuos con alto riesgo quirúrgico, situaciones en las cuales se ha demostrado ampliamente su eficacia y seguridad (66-72).
Las indicaciones clínicas con su respectivo rango de INR recomendado se muestran en la Tabla II (64).
INDICACIONES CLÍNICAS Y VALOR DE INR RECOMENDADO
| INR | Indicación |
| 2,0-2,5 | Profilaxis de trombosis venosa profunda (incluyendo alto riesgo quirúrgico) |
| 2,0-3,0 | Tratamiento de trombosis venosa profunda o embolismo pulmonar Profilaxis de embolismo en pacientes con: – Enfermedad valvular cardíaca – Fibrilación auricular – Prótesis valvulares cardíacas biológicas – Infarto del miocardio con evidencia de trombos murales – Miocardiopatía dilatada con fracción de eyección menor del 25% |
| 2,0-3,5 | Válvulas cardíacas protésicas mecánicas Embolismo sistémico recurrente |
INTENSIDAD DE LA TAO
Un aspecto clave de la TAO lo constituye la aparición de manifestaciones hemorrágicas (aun con valores de INR dentro del rango terapéutico), que conducen a la disminución de la dosis del anticoagulante o a la suspensión temporal del mismo en algunos casos, acarreando como consecuencia durante este período de tiempo, un riesgo elevado de recaída. Debido a esto, ha surgido la tendencia desde hace varios años de utilizar cada vez dosis menores para disminuir el rango terapéutico del INR y lograr de esta manera, un menor índice de complicaciones hemorrágicas, pero claro está, sin aumentar el riesgo de recurrencia trombótica. La finalidad de este esquema es aportar una mejor relación beneficio/riesgo a los pacientes y de igual modo, disminuir los costos de una terapia que puede ser indefinida en muchos casos. Recientemente, dos grandes estudios evaluaron la utilidad de dar dosis bajas de warfarina para mantener el INR entre 1,5 y 1,9 con resultados contradictorios. El estudio PREVENT (Prevention of Recurrent Venous Thromboembolism) (73), logró demostrar que la terapia de baja intensidad a largo plazo es un método efectivo para reducir el riesgo de recurrencia de tromboembolismo. Sin embargo, en el estudio ELATE (Extended Low-Intensity Anticoagulation for Thromboembolism) (74), la tasa de recurrencia de trombosis fue mayor en el grupo de INR entre 1,5 y 1,9 al compararse con el esquema convencional (INR: 2-3), mientras que la incidencia de complicaciones hemorrágicas fue similar.
Pese a estos resultados, el uso de warfarina en forma de esquemas de mini intensidad es una modalidad terapéutica muy atractiva para aquellos pacientes que requieren profilaxis a largo plazo, aunque todavía no esté bien definida su utilidad. No obstante, los pacientes con factores de riesgo trombótico hereditarios (deficiencia de Antitrombina III, PC, Proteína S, presencia del factor V Leiden) o adquiridos (Síndrome Antifosfolipídico, cáncer, entre otros), que tengan un mayor riesgo de trombosis, podrían requerir de un esquema convencional, es decir dosis más altas, para evitar las recaídas.
En conclusión, la estratificación del riesgo de trombosis y hemorragia es fundamental a la hora de seleccionar cual esquema de anticoagulación sería el más beneficioso, especialmente cuando la terapia debe ser extendida indefinidamente.
DURACIÓN DE LA TAO
En la actualidad el tiempo de duración óptimo de la TAO cuando se trata de una trombosis idiopática o cuando el factor de riesgo es perenne, es controversial. Pinede y col. (75), en un metanálisis realizado en el año 2000, para evaluar la duración óptima de la terapia con warfarina luego de un primer evento trombótico venoso, encontraron un riesgo relativo de recurrencia de 0,6 (95% IC: 0,45 a 0,79) cuando la terapia se administró durante tres meses o más comparada con seis semanas o menos de duración. En el año 2001, Pinede y col. (76) compararon los regímenes terapéuticos de tres y seis meses, en trombosis venosas proximales, y de seis semanas y tres meses, en trombosis venosas distales. Si bien la duración de la terapia no influyó en la tasa de recurrencia trombótica ni de hemorragias, aquellos individuos que tenían un factor de riesgo permanente tuvieron una tasa de recurrencia de trombosis mayor que los que tuvieron factores de riesgo transitorios. Kearon y col. (77), observaron a lo largo de once meses, a dos grupos de pacientes que recibieron terapia con warfarina durante uno y tres meses respectivamente, debido a una trombosis venosa profunda o embolismo pulmonar causado por un factor de riesgo transitorio, sin encontrar diferencias en las tasas de recurrencia. Aunque estos hallazgos lucen contradictorios, la mayoría de la guías internacionales de anticoagulación recomiendan dar sólo tres meses de terapia oral para las trombosis venosas distales, mientras que para las proximales o embolismo pulmonar, se sugiere al menos tres meses si son debidas a un factor de riesgo transitorio, pero en los casos donde se trate de un factor de riesgo perenne o sean idiopáticas, el tratamiento deberá prolongarse al menos hasta los seis meses. En casos de eventos repetidos, la anticoagulación debe mantenerse indefinidamente (78).
El riesgo de recurrencia trombótica durante el primer año luego de suspender la TAO, es de 10 a 27% cuando ésta se administra por tres meses (76, 79). Aunque esta cifra disminuye a 10% aproximadamente cuando la terapia se administra por seis meses (80), la extensión de la misma por más de este período de tiempo, no disminuye substancialmente el riesgo de recurrencia luego de descontinuarla. La decisión de prolongar la TAO será el resultado de considerar el riesgo de recurrencia, si se suspende ésta y el riesgo de sangrado, si se mantiene.
Sin duda, la duración de la terapia con warfarina va a depender del tipo de trombosis, de su ubicación, de su etiología, de los antecedentes personales y familiares de trombosis y por supuesto, de los factores predisponentes, bien sean transitorios o permanentes.
VARIABILIDAD DE LA RESPUESTA A LA ACCIÓN DE LA WARFARINA
La necesidad de realizar un control periódico de INR para ajustar la dosis de warfarina, se debe precisamente a la gran variabilidad en la respuesta a su efecto anticoagulante. Existen muchos factores que pueden modificar la dosis necesaria para obtener el efecto anticoagulante deseado. Algunos de ellos, pueden ser controlados y la vigilancia tanto del médico como del propio paciente juega un papel preponderante para el adecuado control de esta terapia.
La dieta es uno de los principales factores que modifican la TAO. Debe recomendarse al paciente que disminuya la ingesta de alimentos con alto contenido de VK, ya que estos crean resistencia adquirida a la warfarina y como consecuencia, se necesitarán dosis más altas para lograr la anticoagulación deseada. Entre estos alimentos se encuentran los vegetales de hojas verdes tales como: Espinaca, col, col de Bruselas, coliflor, brócoli, perejil, habas, nabo, acelga, berro, alcachofa, endibia, guisante, lechuga, espárrago, apio y tomate verde. Se sugiere aportar entre 60 y 80 µg/día de VK, de forma constante, para evitar las fluctuaciones en las dosis requeridas del anticoagulante (81). Algunos investigadores han demostrado que dietas que incluyen un consumo de VK superior a 250 µg/día, producen un descenso importante del INR, aumentando el riesgo de embolias o trombosis. El caso contrario ocurre cuando la ingesta es mínima. Esto sugiere la importancia de un aporte controlado de esta vitamina para mantener estable el valor de INR (82, 83). Al igual que el consumo de VK, la ingesta de alcohol especialmente en abundante cantidad, debe ser controlada porque puede afectar las concentraciones plasmáticas de warfarina (84).
Otro factor que influye en la respuesta es la edad del paciente. Es aconsejable que en sujetos mayores de 60 años, se inicie la terapia con dosis más bajas de las recomendadas para personas jóvenes y realizar controles periódicos de INR de manera más estricta (85, 86). Se han propuesto muchas causas que podrían contribuir a la mayor sensibilidad de la población geriátrica. Cambios en el metabolismo y excreción de los fármacos, la púrpura senil y la ingesta de múltiples medicamentos que interfieren con la warfarina, son algunas de las más relevantes.
El descubrimiento de las variantes alélicas del citocromo P-450 (CYP2C9), principal metabolizador de la warfarina, y del complejo de la VKOR, permitió demostrar que los factores genéticos están implicados en la gran variabilidad individual y la mayor tendencia hemorrágica que se observa en algunos individuos anticoagulados (87). Estos polimorfismos son claves para predecir como reaccionarían los individuos a este medicamento. Las dos variantes más importantes con implicaciones clínicas son la CYP2C9*2 (R144C) y CYP2C9*3 (I359L) y los portadores de éstas (metabolizadores lentos), requieren dosis más bajas y están expuestos a un mayor riesgo de complicaciones hemorrágicas si no se hace el ajuste adecuado de la dosis (88). Por otra parte, el gen de la subunidad 1 del complejo de VKORC1 (16p11.2), también presenta polimorfismos que modulan la dosis diaria de warfarina particularmente el VKORC1 C1173T (89, 90). Por todas estas razones, se recomienda actualmente que a los sujetos con mayor riesgo de hemorragias, les sea descartado este tipo de alteración genética antes del inicio del tratamiento.
La interacción medicamentosa (comentada anteriormente) y la presencia de enfermedades concurrentes, juegan también un papel fundamental en la respuesta a este anticoagulante. Entre las patologías destacan las hepatopatías (disminución de la síntesis de factores de la coagulación), los estados hipermetabólicos como el hipertiroidismo (aumento del catabolismo de los factores dependientes de VK), las enfermedades intestinales (disminución de la absorción) y la insuficiencia renal (alteración de la tasa de unión a proteínas plasmáticas).
COMPLICACIONES DE LA TAO
Si el objetivo de la TAO es crear un estado de hipocoagulabilidad, es lógico suponer que la hemorragia sea el efecto adverso que se presente con mayor frecuencia. Las casuísticas reportadas varían mucho, debido a las diferencias en la clasificación del tipo de hemorragia, así como a las divergencias en el rango de INR utilizado para controlar la terapia. Aunque varios factores pueden incidir en la aparición de esta complicación, la dosis administrada, la edad del paciente, la ingesta simultánea de otros medicamentos y los procedimientos quirúrgicos, son sin duda, los de mayor impacto clínico (91). Es evidente que entre mayor sea el rango de INR utilizado para controlar el efecto anticoagulante, mayor es el riesgo de presentarse una hemorragia que obligue al paciente a consultar a su médico. Afortunadamente, la mayoría de estas hemorragias son menores, es decir no ponen en peligro la vida del paciente, y pueden ser tratadas fácilmente.
Cuando las dosis son ajustadas para mantener un INR entre 2 y 3, el riesgo anual de hemorragias mayores es aproximadamente del 3% (91), mientras que otros investigadores han publicado tasas que van del 5 al 9% (92, 93). En contraste, el uso de dosis bajas (para mantener un INR por debajo de 2), conlleva un menor riesgo de hemorragia, lo cual es especialmente útil en los casos donde está indicada una terapia a largo plazo y además, como beneficio adicional, podría no ser necesario un control clínico y de laboratorio tan riguroso. En el caso de hemorragias menores, Mosley y col. (94), describieron una frecuencia de 22% en individuos con terapia a largo plazo y sólo un 1,5% de casos ameritaron hospitalización debido a su gravedad. Si el control de la TAO es realizado en el medio hospitalario o ambulatorio, también influye en la tasa de hemorragias. Los pacientes hospitalizados tienen menor riesgo de sangrar, debido a que los controles son más sucesivos que en los ambulatorios. Pastor y col. (95), encontraron que el 40% de los pacientes ambulatorios sufre de algún tipo de hemorragia, cifra que disminuye bruscamente a 10% en los hospitalizados. Afortunadamente, sólo del 2 al 10% de éstas son graves.
El riesgo de sangrado es particularmente alto en sujetos que usan simultáneamente aspirina u otros antiinflamatorios no esteroideos y en los mayores de 65 años, con historia previa de enfermedad cerebrovascular isquémica, sangrado gastrointestinal o fibrilación auricular (96, 97).
Las hemorragias más comunes observadas en pacientes anticoagulados son: Equímosis, gingivorragias, hematuria microscópica, sangrado gastrointestinal, hipermenorrea y hematomas. Sin embargo, cualquier órgano o sistema del organismo puede verse afectado. Las hemorragias intracraneales y gastrointestinales son las más temidas porque pueden ser fatales en algunos casos. El hematoma subdural es la forma más frecuente de hemorragia intracraneal (98).
La necrosis cutánea es una complicación poco frecuente asociada al uso de anticoagulantes orales. Su incidencia oscila entre 0,01 y 0,1% y es más frecuente en mujeres (99,100). Generalmente, las zonas afectadas contienen abundante cantidad de grasa subcutánea como: Mamas, glúteos, muslos, abdomen, brazos, y piernas. Las lesiones necróticas características aparecen durante los primeros días de iniciada la terapia y se inician como áreas eritematosas múltiples extremadamente dolorosas, que luego evolucionan a equímosis y ampollas hemorrágicas y finalmente a necrosis. Como se mencionó anteriormente, esta complicación ocurre en individuos con deficiencia de PC, especialmente cuando se inicia la terapia con dosis altas y en los casos donde se produce un cese prematuro de la heparina, es decir antes de haberse logrado la inhibición de los factores procoagulantes dependientes de VK por la acción de la warfarina (60).
Debido a su capacidad de atravesar la barrera placentaria, los derivados cumarínicos deben ser empleados con precaución durante el embarazo, ya que pueden originar malformaciones congénitas y aborto o pérdida fetal. El Síndrome de Warfarina Sódica o Embriopatía Warfarínica (EW) como más comúnmente se le conoce, se presenta en el 6,4% de los nacidos vivos de mujeres que recibieron TAO en el primer trimestre del embarazo (101). Se caracteriza por malformaciones óseas, tales como: Hipoplasia nasal, depresión del puente de la nariz y punteado epifisiario (condrodisplasia punctata). La administración de warfarina durante el segundo trimestre se asocia con malformaciones del sistema nervioso central (ceguera, retardo mental, microcefalia, hidrocefalia, hipotonía), aunque la incidencia de estas complicaciones es mucho menor en comparación con la EW (102), razón por la cual el uso de este anticoagulante en las gestantes, sólo está contraindicado en el primer trimestre. Vitale y col. (103) demostraron que la aparición de malformaciones congénitas está relacionada directamente con las dosis del anticoagulante oral que recibe la gestante.
El aborto espontáneo constituye la complicación más frecuente del uso de warfarina en el embarazo. La frecuencia es muy variable y oscila entre el 20 y 30% (101, 104). Las pérdidas fetales, aunque menos comunes que los abortos, deben ser también consideradas a la hora de indicar este anticoagulante en el segundo trimestre del embarazo (104).
Cuando la warfarina se emplea en el tercer trimestre, debe ser descontinuada tres semanas aproximadamente antes de la fecha probable del parto, debido al riesgo de desencadenar hemorragias severas durante éste. Se han publicado casos de muerte neonatal por hemorragia intracraneal, como consecuencia de la administración de anticoagulantes orales hasta el término del embarazo (105).
Está claro que la indicación de warfarina en cualquier trimestre del embarazo conlleva un gran riesgo para el binomio madre-feto, por lo que muchos médicos prefieren utilizar heparina de bajo peso molecular durante toda la gestación, aunque esto implique un mayor riesgo de trombosis. En el puerperio, dado que la warfarina no pasa a la leche materna, no existe contraindicación para su administración.
REVERSIÓN DEL EFECTO ANTICOAGULANTE DE LA WARFARINA
Un efecto anticoagulante excesivo, que en la mayoría de los casos se debe a sobredosis, puede manifestarse a través de un INR prolongado y/o hemorragias y la conducta dependerá de ambos factores. Cuando el INR se ubique fuera del rango establecido según la patología, pero menor de 6 y sin evidencia de sangrado, se suspenderá la terapia durante 24 a 48 horas debiendo ser reiniciada con la mitad de la dosis o la misma que recibía anteriormente. En el caso de INR mayor de 6 es recomendable administrar dosis pequeñas de VK1 (1-3 mg IM o IV). Similar conducta debe ser adoptada con un INR menor de 6 pero con evidencia clínica de hemorragia menor. Las hemorragias mayores requieren una rápida reversión del efecto anticoagulante, por lo que la administración de dosis mayores de VK1 (5-10 mg IM o IV en infusión lenta), infusión de complejos de protrombina (que contienen factor II, IX y X), concentrados de factor VII o en su defecto, la transfusión de plasma fresco congelado, pudieran ser necesarios (63). Las dosis altas de VK1 crean un estado de resistencia a la warfarina que se traduce en un lapso mayor de tiempo para alcanzar posteriormente el rango terapéutico de INR.
RECOMENDACIONES PARA EL PACIENTE
Es importante aconsejar a los pacientes sobre la restricción de alimentos con alto contenido en VK (aporte controlado), así como también evitar la ingesta de fármacos que puedan potenciar o inhibir el efecto de los anticoagulantes orales. Por otra parte, el paciente debe estar atento ante cualquier signo de sangramiento que presente y comunicarlo inmediatamente al médico tratante para que sea éste y no el propio paciente, el que realice el ajuste requerido de la dosis o la suspensión de la misma, de acuerdo al nivel de INR que tenga.
CONCLUSIONES
Muchas décadas han transcurrido desde que fue aprobado el uso clínico de la warfarina como anticoagulante y permanece aún como la droga de elección, en la mayoría de las situaciones que requieran este tipo de terapia a largo plazo, a pesar de sus múltiples desventajas. La experiencia acumulada durante estas décadas ha permitido conocer que para lograr la optimización de la TAO, es necesario que ésta sea controlada en unidades o centros especializados, que incluyan además del médico especialista, un personal de laboratorio calificado y con experiencia en el control de INR. De igual modo, el paciente deberá recibir educación que le permita conocer las consecuencias de un manejo inadecuado de la terapia, acatar las recomendaciones y estar atento ante cualquier evidencia clínica de hemorragias u otra complicación. Una estrecha relación médico-paciente asegurará una adecuada anticoagulación y permitirá elevar la calidad de vida de estos individuos.
La gran cantidad de desventajas asociadas al uso de warfarina ha sido la base para el desarrollo de nuevos fármacos anticoagulantes orales. Esta nueva generación está dirigida a la inhibición de puntos específicos en el mecanismo de la coagulación (a diferencia de los derivados cumarínicos que actúan sobre todos los factores VK dependientes). Estos pueden ser clasificados en dos categorías: Inhibidores directos del factor X e inhibidores de la trombina. No obstante, su alto costo, la ausencia de un antídoto específico, así como de estudios que comparen su eficacia y seguridad con la warfarina y entre ellos mismos (hasta la fecha no han sido aprobados por la FDA), constituyen los principales obstáculos para que en un futuro cercano, estas drogas puedan suplantar a la warfarina como la primera elección en la prevención de eventos tromboembólicos.
REFERENCIAS
1. Schofield FW. A brief account of a disease in cattle simulating haemorrhagic septicaemia due to feeding sweet clover. Can Vet Rec 1922; 3:74-78. [ Links ]
2. Roderick LM. A problem in the coagulation of the blood: “sweet clover disease of cattle”. Am J Physiol 1931; 96:413-425. [ Links ]
3. Campbell HA, Link KP. Studies on the hemorrhagic sweet clover disease. IV: The isolation and crystallization of the hemorrhagic agent. J Biol Chem 1941; 138: 21-33. [ Links ]
4. Campbell HA, Smith WK, Roberts WL, Link KP. Studies on the hemorrhagic sweet clover disease. II: The bioassay of hemorrhagic concentrates by following the prothrombin level in the plasma of rabbit blood. J Biol Chem 1941; 138:1-20. [ Links ]
5. Link KP. The discovery of dicumarol and its sequels. Circulation 1959; 19(1):97-107. [ Links ]
6. O’Reilly RA. Studies of optical enatiomorphs of warfarin in man. Clinical Pharmacol Ther 1974; 16:348-354. [ Links ]
7. Breckenridge A. Oral anticoagulant drugs: Pharmacokinetic aspects. Semin Haematol 1978; 15:19-26. [ Links ]
8. O’Reilly RA. Interaction of the oral anticoagulant drug warfarin and its metabolites with human plasma albumin. J Clin Invest 1969, 48:193-202. [ Links ]
9. Breckenridge A. Oral anticoagulant – the totem and the taboo. Br Med J 1976; 1: 419-423. [ Links ]
10. Lewis RJ, Trager WF, Robinson AJ, Chan KK. Warfarin metabolites the anticoagulant activity and pharmacology of warfarin alcohols. J Lab Clin Med 1973; 81:925-931. [ Links ]
11. Koch-Weser J. Hemorrhagic reactions and drug reactions in 500 warfarin-treated patients. Clin Pharmacol Ther 1973; 14:139. [ Links ]
12. Starr KJ, Petrie JC. Drug interactions in patients on long-term anticoagulant and antihypertensive adrenergic neuron blocking drugs. Br Med J 1972; 4:133-135. [ Links ]
13. Bechtold H, Andrassy K, Jahnchen E, Koderisch J, Koderisch H, Weilemann LS, Sonntag HG, Ritz E. Evidence for impaired hepatic vitamin k1 metabolism in patients treated with N-methylthiotetrazole cephalosporins. Thromb Haemost 1984; 51:358-361. [ Links ]
14. OReally RA. The stereoselective interaction of warfarin and metronidazole. N Engl J Med 1976; 295:354-357. [ Links ]
15. Christenson LK, Skovsted L. Inhibition of drugs metabolism by chloramphenicol. Lancet 1976; 2:1397-1399. [ Links ]
16. Weibert RT, Lorentz SM, Townsend RJ, Cook CE, Klauber MR, Jagger PI. Effect of erythromycin in patients received long-term anticoagulants. Clin Pharmacol 1989; 8:210-214. [ Links ]
17. O´Reilly RA. Stereoselective interaction of trimethoprim-sulfamethoxazole with the separated enantiomorphs of racemic warfarin in man. N Engl J Med 1980; 302:33-35. [ Links ]
18. Woldtvedt BR, Cahoon CL, Bradley LA, Miller SJ. Possible increased anticoagulation effect of warfarin induced by azithromycin. Ann Pharmacother 1998; 32:269-270. [ Links ]
19. Fausa O. Salicylate-induced hypoprothrombinemia. A report of four cases. Acta Med Scand 1979; 188:403-408. [ Links ]
20. Sellers EM, Koch-Weser J. Displacement of warfarin from human albumin by diazoxide and ethacrynic, mefenamic and nalixidic acids. Clin Pharmacol Ther 1970; 11:524-529. [ Links ]
21. Mersfelder T, Steward L. Warfarin and celecoxib interaction. Ann Pharmacother 2000; 34:325-327. [ Links ]
22. Tempero KF, Cirillo VJ, Steelman SL. Diflunisal: A review of pharmacokinetic and pharmacodynamic properties, drug interactions and special tolerability studies in humans. Br J Clin Phamacol 1977; 4:315-365. [ Links ]
23. Ravic M, Johnston A, Turner P, Ferber HP. A study of the interaction between lornoxicam and warfarin in healthy volunteers. Hum Exp Toxicol 1990; 9:413-414. [ Links ]
24. Rhodes RS, Rhodes PJ, Klein C, Sintek CD. A warfarin-piroxicam drug interaction. Drug Intell Clin Pharm 1985; 19:556-558. [ Links ]
25. Schwartz J, Bugianesi K, Ebel D, De Smet M, Haesen R, Larson P, Ko A, Verbesselt R, Hunt T, Lins R, Lens S, Porras A, Dieck J, Keymeulen B, Gertz B. The effect of rofecoxib on the pharmacodynamics and pharmacokinetics of warfarin. Clin Pharmacol Ther 2000; 68:626- 636. [ Links ]
26. Carter SA. Potential effect of sulindac on response of prothrombin time to oral anticoagulants. Lancet 1979; 11:698. [ Links ]
27. Rees A, Dalal JJ, Reid PG, Henderson AH. Dangers of amiodarone and anticoagulant treatment. Br Med J 1981; 282:1756-1757. [ Links ]
28. Gazzaniga A, Stewart D. Possible quinidine induced haemorrhage in a patient on warfarin sodium. N Engl J Med 1969; 280:711. [ Links ]
29. Hansen J, Siersboek-Nielsen E, Skovstedt L. Carbamazepine-induced acceleration of diphenylhydantoin and warfarin metabolism in man. Clin Pharmacol Ther 1971; 12:539-543. [ Links ]
30. Nappi JM. Warfarin and phenytoin interaction. Ann Intern Med 1979; 90:852. [ Links ]
31. Black DJ, Gidal BE, Seaton TL. An evaluation of the effect of fluconazole on the stereoselective metabolism of warfarin. Clin Pharmacol Ther 1992; 51:184. [ Links ]
32. Cullen SI, Catalano PM. Griseofulvin-warfarin antagonism. JAMA 1967; 199: 582-583. [ Links ]
33. O’Reilly RA, Goulart DA, Kunze KL, Neal J, Gibaldi M, Eddy AC, Trager WF. Mechanisms of the stereoselective interaction between miconazole and racemic warfarin in human subjects. Clin Pharmacol Ther 1992; 51:656-657. [ Links ]
34. Scott AK, Park BK, Breckenridge AM. Interaction between warfarin and propranolol. Br J Clin Pharmacol 1984; 17:559-563. [ Links ]
35. O´Reilly RA, Sahud MA, Aggeler PM. Impact of aspirin and chlorthalidone on the pharmacodynamics of oral anticoagulant drugs in man. Ann NY Acad Sci 1971; 179: 173-186. [ Links ]
36. Serlin MJ, Sibeon RG, Mossman S, Breckenridge A, Williams J, Atwood J, Willoughby J. Cimetidine: interaction with oral anticoagulants in man. Lancet 1979; 2:317-319. [ Links ]
37. Sutfin T, Balmer K, Bostrom N, Eriksson S, Hoglund P, Paulsen O. Stereoselective interaction of omeprazole with warfarin in healthy men. Ther Drug Monit 1989; 11:176-184. [ Links ]
38. Braverman S, Marino M. Sucralfate-warfarin interaction. Drug Intell Clin Pharm 1988; 22:913. [ Links ]
39. Lodwick R, McConkey B, Brown AM. Life threatening interaction between tamoxifen and warfarin. Br Med J 1987; 295:1-141. [ Links ]
40. Adachi Y, Yokoyama Y, Nanno T, Yamamoto T. Potentiation of warfarin by interferon. Br Med J 1995; 311:292. [ Links ]
41. Oliver MF, Roberts SD, Hayes D, Pantridge JF, Suzman MM, Bersohn I. Effect of atromid and ethyl chlorophenoxy-isobutyrate on anticoagulant requirements. Lancet 1963; 1:143. [ Links ]
42. Gross I, Brotman M. Hypoprothrombinemia and hemorrhage associated with cholestyramine therapy. Ann Intern Med 1970; 72:95. [ Links ]
43. Trilli LE, Kelley CL, Aspinall SL, Kroner BA. Potential interaction between warfarin and fluvastatin. Ann Pharmacother 1996; 30:1399-1402. [ Links ]
44. Gaw A, Wosornu D. Simvastatin during warfarin therapy in hyperlipoproteinaemia. Lancet 1992; 340:979-980. [ Links ]
45. Ahmad S. Lovastatin. Warfarin interaction. Arch Intern Med 1990; 150:2407. [ Links ]
46. Rawlins M, Smith M. Influence of allopurinol on drug metabolismo in man. Br J Pharmacol 1981; 48:693-98. [ Links ]
47. Acomb C, Shaw, PW. A significant interaction between warfarin and stanozolol. Pharm J 1985; 284:73. [ Links ]
48. Koch-Weser J. Potentiation by glucagon of the hypoprothrombinemic action of warfarin. Ann Intern Med 1970; 72:331. [ Links ]
49. Owens JC, Neely WB, Owen WR. Effect of sodium dextrothyroxine in patients receiving anticoagulants. N Engl J Med 1962; 266:76-79. [ Links ]
50. Bruning PF, Bonfrer JGM. Aminoglutethimide and oral anticoagulant therapy. Lancet 1983; 2:582. [ Links ]
51. Lewis RJ. Effect of barbiturates on anticoagulant therapy. N Engl J Med 1966; 274:110. [ Links ]
52. Morreale A, Janetzky K. Probable interaction of warfarine and acarbose. Am J Health Syst Pharm 1997; 54:1551-1552. [ Links ]
53. Rothstein E. Warfarin effect enhanced by disulfiram. JAMA 1968; 206:1574. [ Links ]
54. Davis JW, Johns LE. Possible interaction of sulphinpyrazone with coumarins. N Engl J Med 1978; 299:955. [ Links ]
55. Borowski M, Furie BC, Bauminger S, Furie B. Prothrombin requires two sequential metal-dependent conformational transitions to bind phospholipid. J Biol Chem 1986; 261:14969-14975. [ Links ]
56. Cranenburg EC, Schurgers LJ, Vermeer C. Vitamin K: the coagulation vitamin that became omnipotent. Thromb Haemost 2007; 98:120-125. [ Links ]
57. Fasco MJ, Hildebrandt EF, Suttie JW. Evidence that warfarin anticoagulant action involves two distinct reductase activities. J Biol Chem 1982; 257:11210-11212. [ Links ]
58. Furie B, Furie BC. Molecular basis of vitamin-k-dependent g carboxylation. Blood 1990; 75:1753-1762. [ Links ]
59. Deykin D, Wessler S, Reimer SM. Evidence of an antithrombotic effect of dicoumarol. Am J Physiol 1960; 199:1161-1164. [ Links ]
60. McGehee WG, Klotz TA, Epteins DJ, Rapaport SI. Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med 1984; 101:59-60. [ Links ]
61. WHO. Expert Committee on Biological Standardization. Thirty-third Report. World Health Organ Tech Rep Sev 1983: 81-105. [ Links ]
62. Loeliger EA, Poller L, Samama M, Thomson JM, Van den Besselaar AM, Vermylen J, Verstraete M. Questions and answers on prothrombin time standardisation in oral anticoagulant control. Thromb Haemost 1985; 54:515-517. [ Links ]
63. O’Reilly RA, Aggeler PM. Studies on coumarin anticoagulant drugs: Initiation of warfarin therapy without a loading dose. Circulation 1968; 38:169-177. [ Links ]
64. Davidson JF, Colvin BT, Barrowcliffe TW, Kernoff PBA, Machin SJ, Poller L, Preston FE, Walker ID, Mibashan RS, Shinton NK. Guidelines on oral anticoagulation. J Clin Pathol 1990; 143:177-183. [ Links ]
65. Deykin D. Warfarin therapy. N Engl J Med 1970; 283:691-694. [ Links ]
66. Hyers TM, Agnelli G, Hull RD, Morris TA, Samama M, Tapson V, Weg J. Antithrombotic therapy for venous thromboembolic disease. Chest 2001; 119:176S-193S. [ Links ]
67. Singer DE. Randomized trials of warfarin for atrial fibrillation. N Engl J Med 1992; 327:1451-1453. [ Links ]
68. Albers GW, Dalen JE, Laupacis A, Manning WJ, Petersen P, Singer DE. Antithrombotic therapy in atrial fibrillation. Chest 2001;119:194S-206S. [ Links ]
69. Stein PD, Alpert JS, Bussey HI, Dalen JE, Turpie AG. Antithrombotic therapy in patients with mechanical and biological prosthetic heart valves. Chest 2001; 119:220S-227S. [ Links ]
70. Cairns JA, Théroux P, Lewis HD, Ezekowitz M, Meade TW. Antithrombotic agents in coronary artery disease. Chest 2001; 119:228S-252S. [ Links ]
71. Smith P, Anersen H, Holme I. The effect of warfarin on mortality and reinfarction after myocardial infarction. N Engl J Med 1990; 323:147-152. [ Links ]
72. Powers PJ, Gent M, Jay RM, Julian DH, Turpie AG, Levine M, Hirsh J. A randomized trial of less intense postoperative warfarin or aspirin therapy in the prevention of venous thromboembolism after surgery for fractured hip. Arch Inter Med 1989; 149:771-774. [ Links ]
73. Ridker PM, Goldhaber SZ, Danielson E, Rosenberg Y, Eby CS, Deitcher SR, Cushman M, Moll S, Kessler CM, Elliott CG, Paulson R, Wong T, Bauer KA, Schwartz BA, Miletich JP, Bounameaux H, Glynn RJ, PREVENT Investigators. Long-term, Low-Intensity Warfarin Therapy for the prevention of recurrent venous thromboembolism. N Engl J Med 2003; 348(15):1425-1434. [ Links ]
74. Kearon C, Ginsberg JS, Kovacs MJ, Anderson DR, Wells P, Julian JA, MacKinnon B, Weitz JI, Crowther MA, Dolan S, Turpie AG, Geerts W, Solymoss S, van Nguyen P, Demers C, Kahn SR, Kassis J, Rodger M, Hambleton J, Gent M, Extended Low-Intensity Anticoagulation for Thrombo-Embolism Investigators. Comparison of low-intensity Warfarin therapy with conventional-intensity Warfarin therapy for long-term prevention of recurrent venous thromboembolism. N Engl J Med 2003; 349:631-639. [ Links ]
75. Pinede L, Duhaut P, Cucherat M, Ninet J, Pasquier J, Boissel JP. Comparison of long versus short duration of anticoagulant therapy after a first episode of venous thromboembolism: a meta-analysis of randomized controlled trials. J Inter Med 2000; 247(5):553-562. [ Links ]
76. Pinede L, Ninet J, Duhaut P, Chabaud S, Demolombe-Rague S, Durieu I, Nony P, Sanson C, Boissel JP, Investigators of the “Durée Optimale du Traitement AntiVitamines K” (DOTAVK) Study. Comparison of 3 and 6 months of oral anticoagulant therapy after a first episode of proximal deep vein thrombosis or pulmonary embolism and comparison of 6 and 12 weeks of therapy after isolated calf deep vein thrombosis. Circulation 2001; 103(20):2453-2460. [ Links ]
77. Kearon C, Ginsberg J, Anderson D, Kovacs M, Wells P, Julian J, Mackinnon B, Demers C, Douketis J, Turpie A, Van Nguyen P, Green D, Kassis J, Kahn S, Solymoss S, Desjardins L, Geerts W, Johnston M, Weitz J, Hirsh J, Gent M. Comparison of 1 month with 3 months of anticoagulation for a first episode of venous thromboembolism associated with a transient risk factor. J Thromb Haemost 2004; 2:743-749. [ Links ]
78. British Committee for Standards in Haematology. Guidelines on oral anticoagulation (warfarin): third edition- 2005. [ Links ]
79. Agnelli G, Prandoni P, Santamaría M, Bagatella P, Bazzan M, Moia M, Guazzaloca G, Bertoldi A, Tomasi C, Scannapieco G, Ageno W. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. N Engl J Med 2001; 345:165-169. [ Links ]
80. Baglin T, Luddintong R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet 2003; 362:523-526. [ Links ]
81. Couris RR, Tataronis GR, Booth SL, Dallal GE, Blumberg JB, Dwyer JT. Development of a selfassessment instrument to determine daily intake and variability of dietary vitamin K. J Am Coll Nutr 2000; 19(6):801-806. [ Links ]
82. Lubetsky A, Dekel-Stern E, Chetrit A, Lubin F, Halkin H. Vitamin K intake and sensitivity to warfarin in patients consuming regular diets. Thromb Haemost 1999; 81(3):396-399. [ Links ]
83. Sorano GG, Biondi G, Conti M, Mameli G, Licheri D, Marongiu F. Controlled vitamin K content for improving the management of poorly controlled anticoagulated patients: a clinical practice proposal. Haemostasis 1993; 23(2):77-82. [ Links ]
84. Cropp J, Bussey H. A review of enzyme induction of warfarin metabolism with recommendations for patients management. Pharmacotherapy 1997; 17(5):917-928. [ Links ]
85. Casner P, Sandoval E. Increased sensitivity to warfarin in elderly hispanics. J Clin Pharmacol 2002; 42:145. [ Links ]
86. Wynne H, Cope L, Kelly P, Whittingham T, Edwards C, Kamali F. The influence of age, liver size and enantiomer concentrations on warfarin requirements. Br J Clin Pharmacol 1995; 40(3):203-207. [ Links ]
87. Takahashi H, Echizen H. Pharmacogenetics of CYP2C9 and interindividual variability in anticoagulant response to warfarin. Pharmacogenomics J 2003; 3: 202-214. [ Links ]
88. Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: A HuGEnet™ systematic review and meta-analysis. Genet Med 2005; 7(2):97-104. [ Links ]
89. Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, Blough DK, Thummel KE, Veenstra DL, Rettie AE. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352(22):2285-2293. [ Links ]
90. D’Andrea G, D’Ambrosio RL, Di Perna P, Chetta M, Santacroce R, Brancaccio V, Grandone E, Margaglione M. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood 2005; 105(2):645-649. [ Links ]
91. Linkins LA, Choi PT, Daeketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893-900. [ Links ]
92. Fihn SD, Callahan CM, Martin DC, McDonell MB, Henikoff JG, White RH. The risk for and severity of bleeding complications in elderly patients treated with warfarin. Ann Intern Med 1996; 124:970-979. [ Links ]
93. McMahan DA, Smith DM, Carey MA, Zhou XH. Risk of major hemorrhage for out-patients treated with warfarin. J Gen Intern Med 1998; 13:311-316. [ Links ]
94. Mosley DH, Schatz IJ, Breneman GM, Keyes JW. Long-term anticoagulant therapy: Complications and control in a review of 987 cases. JAMA 1963; 186: 914-916. [ Links ]
95. Pastor BM, Resnick ME, Rodman T. Serious hemorrhagic complications of anticoagulant therapy. JAMA 1962; 180:747-751. [ Links ]
96. Graham DY, Smith JL. Aspirin and the stomach. Ann Intern Med 1986; 104:390-398. [ Links ]
97. Hirch J. Oral anticoagulant drugs. N Engl J Med 1991; 324: 1865-1874. [ Links ]
98. Silverstein A. Neurological complications of anticoagulant therapy. A neurologist’s rewiew. Arch Intern Med 1979; 139:217-220. [ Links ]
99. Chan Y, Valentin D, Mansfield A. Warfarin induced skin necrosis. Br J Surg 2000; 87:266-272. [ Links ]
100. Cole M, Minifee P, Wolman F. Coumarin necrosis-a review of the literature. Surgery 1988; 103:271-277. [ Links ]
101. Chan WS, Anaud S, Gingsberg JS. Anticoagulation of pregnant women with mechanical heart valves: a systematic review of the literature. Arch Intern Med 2000; 160:191-196. [ Links ]
102. Hall JG, Pauli RM, Wilson KM. Maternal and fetal sequelae of anticoagulation during pregnancy. Am J Med 1980; 68:122-140. [ Links ]
103. Vitale N, De Feo M, De Santo, Pollice A, Tedesco N, Cotrufo M. Dose-dependent fetal complications of warfarin in pregnant women with mechanical heart valves. J Am Coll Cardiol 1999; 33:1637-1641. [ Links ]
104. Salazar E, Izaguirre R. Heart disease, anticoagulants and pregnancy. Rev Esp Cardiol 2001; 54(Supl.1):8-16. [ Links ]
105. Sareli P, England MJ, Berk MR, Marcus RH, Epstein M, Driscoll J, Meyer T, McIntyre J, van Gelderen C. Maternal and fetal sequelae of anticoagulation during pregnancy in patients with mechanical heart valve prostheses. Am J Cardiol 1989; 63:1462-1465. [ Links ]

