










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.19 n.2 Caracas jul. 2000
Aspectos clínicos y terapéuticos de las Trombosis Venosas y Arteriales
M Rivera1, F Contreras1, M de la Parte1, O Méndez2, Y Colmenares2 y M Velasco3.
- Departamento de Ciencias Básicas, Cátedra de Fisiopatología EEE, Facultad de Medicina – UCV.
- Departamento de Medicina Interna HVSR
- Unidad de Investigación Cardiovascular Vargas, Escuela de Medicina "J.M Vargas" Facultad de Medicina UCV , Caracas.
RESUMEN
La integridad del endotelio vascular desempeña un papel crucial en el mantenimiento de la fluidez sanguínea al igual que las sustancias expresadas y liberadas por las células endoteliales y sus membranas. La trombosis venosa y arterial son trastornos comunes, potencialmente letales y afectan por igual a todas las etnias. Estos padecimientos aumentan progresivamente con la edad y se tornan comunes en pacientes hospitalizados. La heparina a bajas dosis se emplea como medida profiláctica de la Trombosis Venosa Profunda (TVP), y a dosis altas se utiliza cuando la patología implica un riesgo trombótico mayor, ya sea arterial o venoso; las Heparinas de Bajo Peso Molecular (HBPM) son ideales para la prevención de TVP. Los anticoagulantes orales constituyen agentes muy importantes en el arsenal terapéutico contra los fenómenos trombóticos, independientemente de su etiología. La Aspirina es el agente más estudiado y usado de los antiagregantes plaquetarios; se ha utilizado en el control y evaluación de otros agentes antiplaquetarios como ticlopidina, clopidogrel, aspirina más dipiridamol de liberación prolongada, actualmente recomendados. Como alternativa y para evitar la trombocitopenia inducida por la heparina, tenemos en estudio los inhibidores de la trombina y se esperan resultados alentadores. En relación con el tratamiento trombolítico, éste se debe instaurar en las primeras horas después de realizado el diagnóstico, teniendo en cuenta sus indicaciones y contraindicaciones, siendo cuidadosos al escoger el agente y que éste sea manejado por manos expertas, en centros adecuados y en aquellos casos que así lo ameriten; siempre debe tenerse presente el riesgo de hemorragia como efecto adverso con el uso de cualquiera de estos agentes.
Palabras Claves: Trombosis arterial, Trombosis venosa, Anticoagulantes, Heparinas.
ABSTRACT
The integrity of the vascular endothelium is of primary importance for the maintenance of blood fluidity as well as the substances expressed and released by endothelial cells and membranes. Venous and arterial thrombosis are common diseases potentially fatal affecting equally to all races. These ailments are more frequent with increasing age and there are common in hospitalized patients. Heparin at low dosage is being used as preventive for Deep Vein Thrombosis (DVT); higher doses are given in pathologies with a higher risk of venous or arterial thrombosis; Low Molecular Weight Heparins (LMWH) are ideal for prevention of DVT. Oral anticoagulants are a very important therapeutic group against events of thrombosis regardless of ethiology. Aspirin has been the most studied and used molecule to prevent platelet aggregation; actually is recommended at low doses and as been compared with other products used at present for the prevention of platelet aggregation such as ticlopidin, clopidrogrel and slow liberation of aspirin combined with dipiridamol. To overcome the heparin-induced thrombocytopenia there are being tryed thrombin inhibitors with great hope in good results. As for the thrombolytic treatment itself, it should be initiated during the first hours of diagnosis keeping in mind indications and counterindications, being very careful in the choice of the agent, used by experts, in those cases where necessary and in patients in hospitals with facilities for adequate control. It should also be kept in mind the risk of hemorrhage as side effect in the therapeutic with these agents.
Key Words: Arterial thrombosis, Venous thrombosis, Anticoagulants, Heparins.
INTRODUCCION
La formación del trombo venoso está dada principalmente por la activación de la coagulación de la sangre y el éstasis venoso. En tanto que las anormalidades vasculares son el factor principal en la trombosis arterial.
La trombosis venosa profunda (TVP) es un trastorno común. Es más frecuente en las mujeres que en los varones: todas las etnias están afectadas por igual. El padecimiento se vuelve más frecuente conforme aumenta la edad y es común en pacientes inmovilizados. Se presenta en casi la tercera parte de los pacientes mayores de 40 años, sometidos a cirugía mayor o después de un infarto de miocardio. La frecuencia es aún mayor después de ciertas operaciones, especialmente en la cirugía ortopédica requerida por pacientes con fracturas de cadera o urológicas como prostatectomías. La TVP también es muy elevada en pacientes con accidentes cerebrales vasculares de tipo trombótico. Los factores etiológicos asociados a la TVP, pueden ser divididos en factores clínicos y defectos plasmáticos hereditarios o adquiridos(1,2). Las condiciones clínicas relacionadas a TVP son: éstasis venosa, septicemia, postoperatorio, neoplasias, traumatismos, insuficiencia cardiaca congestiva, síndrome nefrótico, insuficiencia venosa, obesidad, quemaduras, edad avanzada, uso de estrógenos y problemas anatómicos vasculares.
Las alteraciones hematológicas que predisponen a fenómenos trombóticos son: deficiencia de antitrombina III, proteína C, proteína S, plasminógeno, cofactor II de heparina, activador tisular del plasminógeno (t-PA), deficiencia de cistionina B sintetasa, desfibrinogenemia, el recientemente descrito defecto molecular del factor V resistente a la proteína C activada, policitemia, presencia de anticoagulante lúpico o anticuerpos antifosfolípidos, trombocitosis malignas y benignas. La deficiencia de factor XII, elevación del factor VII, fibrinógeno y deficiencia de lipoproteína A, pueden también estar asociados a estados de hipercoagulabilidad. La literatura ha demostrado que el 40% de los casos de trombosis juvenil o recurrente, se asocian a resistencia a la proteína C activada y el 10% se relacionan a deficiencia de proteína S, C, antitrombina III y otras condiciones raras. Considerando las trombosis venosas de forma general, apenas el 20% está relacionado a estas causas(3,4,5). Se ve, aunque con menor frecuencia, que los defectos plaquetarios pueden también originar fenómenos trombóticos arteriales y venosos. (Ej.: Síndrome de plaquetas pegajosas)
Por otra parte, la ruptura de una placa ateroesclerótica en una arteria epicárdica origina una secuencia de eventos que conllevan a la formación de un trombo local que, dependiendo del grado de oclusión podrá desencadenar una isquemia o necrosis en el área de miocardio suplido por esa arteria(6) . En la mayoría de los casos, la angina inestable se debe a la formación de un trombo mural o a la ruptura o erosión de una placa ateroesclerótica; sin embargo, cualquier proceso que en forma aguda produzca cambios en la relación oferta-demanda (disminución de la oferta o incremento de la demanda en presencia de una disminución en la oferta) pueden precipitar la presentación clínica de una angina inestable o más allá, un infarto de miocardio (7).
Los fenómenos tromboembólicos han sido objeto de múltiples estudios en los últimos años, teniendo como objetivos: identificar condiciones predisponentes, establecer estrategias diagnósticas más precisas y de menor costo y finalmente identificar terapias de menor riesgo y mayor efectividad; de esto último, nos ocuparemos en este artículo.
REOLOGIA DE LA SANGRE
La reología es una disciplina que se ocupa de flujos y deformaciones de materiales sometidos a la acción de fuerzas mecánicas. Las propiedades físicas del flujo sanguíneo dependen de parámetros tales como la viscosidad sanguínea, la viscosidad plasmática, la deformabilidad de los glóbulos rojos y la agregación eritrocitaria; todos estos factores juegan un papel importante en los mecanismos fisiopatológicos que conducen a la formación de la placa de ateroma, en la hemostasia, la trombosis y la vasomotricidad vascular.
La viscosidad plasmática depende de la concentración de proteínas plasmáticas y particularmente de macromoléculas como el fibrinógeno. Numerosos estudios clínicos y epidemiológicos demuestran que una tasa de fibrinógeno plasmático elevada es un factor de riesgo independiente de enfermedad coronaria, cerebro-vascular, de las arterias de los miembros inferiores y del sistema venoso.
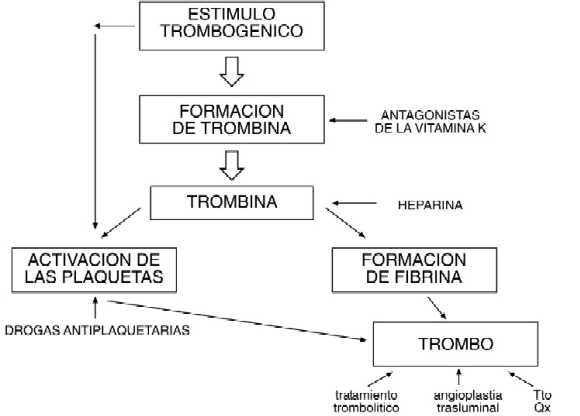
Fig 1-1: Sitio de acción de los principales agentes Antiplaquetarios, Trombolíticos y Antitrombolíticos.
¿QUÉ ES LO QUE MANTIENE LA FLUIDEZ DE LA SANGRE?
Diversos experimentos han señalado la importancia del revestimiento endotelial para el mantenimiento de la fluidez de la sangre. Los avances efectuados en la última década, han aportado datos sólidos que apoyan las primeras observaciones, y a nivel molecular los datos obtenidos han empezado a aclarar los mecanismos bioquímicos. En ausencia de un trastorno significativo, la fluidez de la sangre se mantiene por el flujo continuo y por las cargas negativas de las superficies celulares; ya que si el vaso sanguíneo se mantiene intacto no ocurre activación plaquetaria, ni coagulación. Quizá más importantes son las sustancias expresadas en la membrana externa de la célula endotelial (trombomodulina, heparinas, ADPasa) o liberadas por las células endoteliales (PGI2, EDRF y t-PA) las cuales actúan minimizando la activación de las plaquetas y la coagulación y, en el caso de la formación de fibrina, eliminando o destruyendo los trombos (8).
El papel que desempeña el endotelio vascular en el mantenimiento de la fluidez de la sangre en ausencia de alteraciones importantes se realiza a través de varias moléculas que están situadas en la superficie de las células endoteliales o bien son liberadas por estas células. Entre ellas están la prostaciclina (PGI2), el óxido nítrico (NO), que inhibe la adhesión plaquetaria. Al igual que la ADPasa, la PGI2 y el NO también inhiben la activación y agregación plaquetarias. La trombomodulina y las moléculas de tipo heparínico inhiben la coagulación y el activador hístico del plasminógeno(t-PA) activa la fibrinólisis(8) .
TRATAMIENTO DE LOS EVENTOS TROMBOTICOS
Los medicamentos que modifican directa o indirectamente la cinética de la coagulación sanguínea son las heparinas, los dicumarínicos, los antiagregantes plaquetarios y los agentes trombolíticos(2). Además, de los agentes antitrombina actualmente en estudio. Algunas de estas drogas actúan potenciando los efectos de los anticoagulantes naturales, los cuales permiten que la coagulación se lleve a cabo localmente en respuesta a la lesión y evitan que se vuelva un proceso sistémico y potencialmente letal. Los tres anticoagulantes naturales más importantes son: antitrombina III, proteína C y proteína S (9).
HEPARINA NO FRACCIONADA
En 1.916, un estudiante de medicina, mientras estudiaba la naturaleza de los procoagulantes solubles en éter, halló en forma accidental un anticoagulante fosfolipídico; poco después, en 1.922, Howell, descubrió un mucopolisacárido soluble en agua llamado heparina por su abundancia en hígado. El uso de heparina «in vitro» para prevenir la coagulación de la sangre, llevó finalmente a su empleo «in vivo» para el tratamiento de la trombosis venosa (10).
La heparina es una mezcla heterogénea de polímeros de un polisacárido natural, extraído de vísceras animales, con peso molecular que varía de 3.000 a 30.000 daltons. Una interacción con la antitrombina III le confiere su principal efecto anticoagulante, a través de un cambio en la conformación en la misma, que acelera su habilidad en inactivar la trombina y los factores Xa y IXa (2).
La actividad anticoagulante de la heparina no fraccionada depende de un pentasacárido único que se une a la antitrombina III y potencia la inhibición de la trombina y del factor X activado (Xa) por la antitrombina III(11,12,13). Para la inhibición de la trombina, es necesario que la heparina forme un puente entre la trombina y la antitrombina III, pero para la inhibición del factor Xa, ésto no es preciso (14,15). La heparina no fraccionada es incapaz de inhibir la trombina unida a fibrina, tampoco inhibe el factor Xa unido a plaquetas (15). La heparina también cataliza la inactivación de trombina por otro cofactor plasmático (cofactor II), que actúa de modo independiente sobre la antitrombina III. La heparina tiene otros efectos. Los relacionados con los efectos anticoagulantes incluyen la liberación del inhibidor de la vía del factor hístico, unión a muchas proteínas plasmáticas y plaquetarias, células endoteliales y leucocitos, supresión de la función plaquetaria, y aumento de la permeabilidad vascular (16).
La respuesta anticoagulante a una dosis estándar de heparina varía mucho entre los pacientes. La depuración plasmática de la heparina depende de una depuración renal relacionada con la dosis y un mecanismo celular saturable dosis independiente(17). La unión de la heparina a las proteínas plasmáticas, células endoteliales, y plaquetas contribuye a la respuesta impredecible(18,19). Algunos individuos presentan resistencia relativa a la heparina y necesitan una dosis grande para alcanzar respuesta en el tiempo parcial de tromboplastina activada (TTPa). La respuesta anticoagulante de la heparina se debe vigilar con el TTPa o con las cifras de la heparina y se titulará la dosis de cada paciente en forma individualizada.
La heparina puede ser utilizada en esquemas terapéuticos con bajas o altas dosis. Su utilización en bajas dosis, está indicada cuando se desea prevenir la TVP en pacientes con factores de riesgo trombóticos (20). Las altas dosis son utilizadas con fines terapéuticos cuando se pretende prevenir la ocurrencia de un segundo episodio tromboembólico en una TVP ya instalada, en pacientes con TVP, embolismo pulmonar, trombosis o embolias arteriales, pacientes sometidos a procedimientos que implican un riesgo trombótico, como cateterismos arteriales o angioplastias, hemodiálisis o cirugía cardiovascular con circulación extracorpórea .
El beneficio del uso profiláctico con bajas dosis de heparina está mediado por el efecto antitrombótico de la antitrombina III relacionada al factor Xa. Este factor, es importante tanto para el sistema intrínsico de la coagulación como para el extrínsico e influye también en la agregación plaquetaria. La acción fibrinolítica de la heparina es discutida, y probablemente sea indirecta por la reducción de la actividad coagulante. Las dosis utilizadas son de 5.000 a 7.500 UI de heparinato de calcio o de sodio cada 8 o 12 horas, por vía subcutánea. En el caso de cirugías, la profilaxis debe iniciarse antes del acto operatorio. Las alteraciones de laboratorio, sobre todo el tiempo de tromboplastina parcial activado (TTPa) son insignificantes o inexistentes y las complicaciones hemorrágicas intra o postoperatorias raramente son significativas. La realización de controles de TTPa 2 a 5 horas después de administrada la heparina, puede ser útil para detectar pacientes hiperreactivos, absorción brusca del medicamento y error en la administración (VIM en lugar de SC) (21).
La utilización de la heparina a altas dosis puede ser administrada por vía intravenosa o SC, siendo la primera la preferida, por producir un efecto inmediato y no producir hematomas locales. La administración EV puede ser intermitente o continua. Con la infusión continua se administran dosis diarias más bajas, controles más adecuados y menor incidencia de complicaciones hemorrágicas. Dependiendo de la condición clínica del paciente debe aplicarse una dosis de ataque, de 2.500 a 5.000 UI, seguido de un esquema de mantenimiento por infusión continua que dependerá de los controles de laboratorio, pero usualmente varía entre 1.000 a 2.000 UI/h (20). En los casos de coagulación intravascular diseminada, cuando el recuento plaquetario es bajo, la dosis no debe sobrepasar 500 UI/h (2).
La complicación más grave con el uso de la heparina es la hemorragia, relacionada en primer lugar con los factores de riesgo clínicos fundamentales pero también esta aumentada en mujeres e individuos de más de 65 años de edad(22). La segunda complicación importante y bien identificada es la trombocitopenia inducida por la heparina(23,24,25) que por lo general ocurre en el transcurso de 5 a 10 días después de iniciada la terapia(26,27,28). Es posible que la trombocitopenia se acompañe de trombosis arterial o venosa, que puede llevar a graves consecuencias como amputación de extremidades o muerte. El diagnóstico de esta complicación, con trombosis o sin ella, aunque es difícil, es eminentemente clínico.
También, se ha señalado osteoporosis en individuos que reciben heparina no fraccionada en dosis de 20.000 U/día (o más) durante más de seis meses(29). Entre otras complicaciones atribuidas a la heparina tenemos: aumento en la concentración de las enzimas hepáticas, hipoaldosteronismo, hipersensibilidad y reacciones cutáneas alérgicas, así como necrosis cutánea inducida por heparina(29).
HEPARINAS DE BAJO PESO MOLECULAR
Las heparinas de bajo peso molecular (HBPM) se obtienen por despolimerización química o enzimática de la heparina no fraccionada (HNF). Las HBPM empleadas clínicamente tienen un peso molecular medio de 4.000 a 6.000 daltons (12), comparado con 15.000 daltons que es el peso molecular medio de la HNF. Son más débiles que la HNF como inhibidoras de la trombina (factor II a), pero su grado de acción es similar al de la HNF como inhibidoras de la enzima Xa de la coagulación. Por ende, la razón de actividad anti-Xa de las HBPM es de dos a cuatro veces mayor que la de las HNF. La actividad anti-Xa parece ser un factor determinante de la actividad anticoagulante de las HBPM(30,31).
Después de la inyección subcutánea (SC), las HBPM de cadena más corta se absorben mejor que la HNF y se ligan menos a las proteínas del plasma y de la pared endotelial(32). Como resultado, su biodisponibilidad, medida por la actividad anti-Xa en el plasma es de cerca del 90% en todas las dosis subcutáneas, mientras que la de la HNF aumenta con la dosis y es solamente de alrededor de 10-30% en dosis profilácticas(32,33,34). Después de la inyección SC, el período de semi eliminación plasmática de las HBPM es de unas 4 horas, lo suficiente para lograr una acción anticoagulante eficaz con una sola inyección diaria en las dosis actualmente recomendadas; el de la HNF es de 1 hora y media, de manera que ésta debe administrarse más frecuentemente. La variación del efecto anticoagulante de la HNF entre una persona y otra y en cada una de ellas exige un monitoreo regular para ajustar la dosis terapéutica, lo cual no es necesario con las HBPM.
Las HBPM se eliminan predominantemente por los riñones, por lo que su concentración plasmática puede elevarse en la insuficiencia renal(35). Las características moleculares ya discutidas otorgan a las heparinas de bajo peso molecular un perfil de fármaco capaz de evitar la hiperactivación del sistema de coagulación sin aumentar el riesgo de sangramiento; esta propiedad hace que estas drogas sean agentes antitrombóticos ideales para prevenir la trombosis venosa profunda posquirúrgica(36).
Diversos estudios controlados demuestran la eficiencia del uso de las HBPM en la prevención del tromboembolismo por cirugías generales u ortopédicas en pacientes de alto riesgo trombótico. Aparentemente, la utilización de una aplicación diaria de 7.500 unidades anti-Xa, iniciándose la primera dosis en el preoperatorio es más eficiente que la utilización exclusivamente postoperatoria. Algunos autores afirman que, que cuando se utiliza la HBPM solo en el postoperatorio, la dosis debe ser de 15.000 UI/ día (36).
La fraxiparina a una dosis de 450U/Kg/día y la enoxiparina a una dosis de 2 mg/Kg/dia, administrada una o dos veces al día, han demostrado mayor eficiencia y seguridad que el régimen de HNF. La eficiencia ha sido evaluada a través de los cambios en el tamaño de los trombos, menor recurrencia de tromboembolismo y disminución de la mortalidad. La disminución de la incidencia de complicaciones hemorrágicas con las HBPM sugieren que puede ser posible el tratamiento ambulatorio de casos seleccionados de TVP(36) . El riesgo de trombocitopenia es menor con las HBPM que con la HNF.
En un estudio aleatorio36, la enoxaparina, a dosis fijas de 30 mg SC cada 12 horas, demostró mayor efectividad que la warfarina en la reducción de trombosis venosas profundas en enfermos intervenidos de prótesis total de rodilla(36).
En el caso de los síndromes coronarios agudos, el tratamiento con heparinas de bajo peso molecular, administradas SC una vez al día, ha mostrado buena efectividad y menores efectos secundarios que con la administración convencional de heparina no fraccionada en infusión continua(37,38) y cuando se asocia con inhibidores plaquetarios glicoproteicos IIb/IIIa son aún más efectivas(38) .
Las diversas HBPM difieren en términos del peso molecular medio, el contenido de C-glucosaminoglucanos, la actividad anticoagulante en términos de actividad anti-Xa y anti-Iia (tabla 1-4). Las diversas fracciones muestran perfiles farmacológicos diferentes en términos de biodisponibilidad, depuración plasmática y liberación de inhibidor de la vía del factor hístico, y en modelos experimentales tienen propiedades antitrombóticas y hemorrágicas diferentes(39,40,41,42,43,44,45). Aunque existen varias diferencias entre las HBPM, los resultados de los estudios clínicos son similares, especialmente en estudios profilácticos mediante dosis más bajas. A dosis más altas es posible que queden manifiestas diferencias en los resultados.
Se están acumulando pruebas de que las complicaciones con la HBPM son menos graves y menos frecuentes que con la HNF. La HBPM no ha sido aprobada para la prevención o terapéutica de tromboembolia venosa durante el embarazo, aunque estas moléculas no cruzan la placenta(46,47). El tratamiento actual estándar para la tromboembolia venosa durante el embarazo consiste en administrar HNF por vía subcutánea en dosis ajustadas, dos veces al día(48).Es importante mencionar que todas las HBPM tienen reacción cruzada con la HNF, por lo que no se deben usar en pacientes con trombocitopenia inducida por heparina.
Tabla 1-1 Principales Contraindicaciones de la Terapia Anticoagulanteraindicaciones de la Terapia Anticoagulante
| ABSOLUTAS | RELATIVAS |
| Hemorragia activa | Hipertensión arterial severa, edad avanzada |
| Trauma reciente del SNC | Endocarditis bacteriana |
| Hemorragia reciente del SNC | Sangramiento digestivo reciente |
| Coagulopatía grave | Retinopatía diabética |
| Hipersensibilidad a la droga | Trombocitopenia grave (<100.000/mm3) |
ANTICOAGULANTES ORALES
El descubrimiento de los anticoagulantes orales es realizado por hallazgos fortuitos y observación cuidadosa. Dos patólogos veterinarios, Schofield(49) y Roderick(50), identificaron por primera la enfermedad hemorrágica de ganado vacuno que se creía era consecuencia de la ingestión de trébol cloroso contaminado. Roderick logró demostrar una deficiencia de protrombina, una proteína dependiente de la vitamina K, en ese ganado vacuno afectado(50). En 1.935, Quick(51) describió el tiempo de protrombina (PT); interesado en explicar el porque de las complicaciones hemorrágicas en pacientes con hepatopatía. Creó el tiempo de protrombina como prueba para valorar la función hepática. En 1.939, Link(52,53) aisló el compuesto dicumarol. A partir de su trabajo continuo con compuestos tipo cumarina, creó así el fármaco (warfarina sódica). A principios de 1.950, la warfarina sódica se introdujo como un anticoagulante para uso en seres humanos. Esta categoría incluye también los derivados de la indadiona. A pesar de su uso frecuente en todo el mundo, existen aun diversos puntos de controversia. El mecanismo de acción de estas sustancias es debido a su semejanza química con la vitamina K1, con la cual compiten en la fase final de la síntesis de los factores II, VII, IX, y X y de las proteínas C y S; en realidad provocan la aparición de formas no carboxiladas de estos factores, incapaces de actuar adecuadamente en la cinética de la coagulación (36).
El efecto de los anticoagulantes orales no es inmediato y depende de varios factores(10,54) entre ellos el apego a la prescripción, la dieta, variaciones en el aporte de vitamina K en la alimentación, hepatopatía subyacente, consumo de alcohol, edad y trastornos gastrointestinales(55,56,57). Se debe tener en cuenta que las interacciones con otros fármacos pueden disminuir o aumentar la respuesta a la warfarina. El tipo de droga utilizada, de acción corta, intermedia y prolongada. La sensibilidad a los anticoagulantes varía mucho de un sujeto a otro, por lo que la dosis debe ser ajustada individualmente.
Las contraindicaciones absolutas para anticoagulantes orales incluyen falta de apego del enfermo a la prescripción, hepatopatía grave e intervención quirúrgica reciente del sistema nervioso central o del globo ocular.
Para iniciar el tratamiento de una TVP, hoy día, los pacientes reciben tanto warfarina como heparina en el momento del diagnóstico, administrándose, la primera con una dosis de saturación de 10 mg. Se obtienen tiempos de protrombina subsecuentes hasta que el International Normalized Ratio (INR) esté dentro del limite blanco de 2 a 3. Posterior al egreso del paciente del hospital durante la primera semana se verifica el INR y una vez que éste se hace estable están indicadas valoraciones repetidas aproximadamente cada 4 a 8 semanas (58).
La hemorragia es la complicación más frecuente del uso de anticoagulantes orales, bien sea por falta de monitoreo regular o por el uso de drogas potenciadoras de los efectos anticoagulantes. Si el INR está dentro del limite terapéutico, se debe excluir una causa subyacente (enfermedad maligna gastrointestinal, úlceras). En caso de hemorragia que ponga en peligro la vida, puede estar indicada la administración de vitamina K, plasma fresco congelado o concentrados de complejos de protrombina.
En cuanto a los derivados de la indadiona, la anisindiona, disponible en Estados Unidos, tiene acción similar a la warfarina aunque no ofrece ventajas claras y puede causar efectos adversos con mayor frecuencia. La fenindiona fue muy utilizada y aun existe; puede ocasionar reacciones de hipersensibilidad graves al poco tiempo de iniciado el tratamiento por lo que no se recomienda su uso actual.
Lugares de acción y posibles efectos de la formación de Plasmina
durante el tratamiento Trombólitico.
| Sitios de acción | Acciones fisiológicas | Efectos clínicos |
| Plasma | Inactivación de factores V y VIII | Hipercoagulabilidad |
| Fibrinólisis | Alteración de la hemostasia | |
| Acúmulo de productos de degradación del fibrinógeno | ||
| Fibrina | Fibrinólisis | Trombolisis |
| Acúmulo de productos de degradación de fibrina | Lisis del tapón hemostático y hemorragia | |
| Membranas Plaquetarias | Degradación del receptor de las glucoproteínas Ib, IIb/IIIa en la membrana plaquetaria | Alteración de la adhesión y agregación plaquetaria Alteración de la hemostasia |
| Matriz interplaquetaria | Degradación de la trombospondina, fibronectina fibrinógeno, fibrina y glucoproteína rica en histidina, | Desagregación de plaquetas Lisis de los tapones hemostáticos y hemorragia |
Tabla 1-3: Agentes Trombolíticos de uso Frecuente
| Estreptocinasa: | Se combina con el plasminógeno o plasmina activando el plasminógeno. Se produce en cepas de Estreptococos Beta-hemolíticos |
| Complejo plasminógeno activador de la estreptocinasa | Formado de la unión de la estreptocinasa con el plasminógeno. Presenta especificidad por la fibrina. |
| Activador hístico del plasminógeno | Producido por los tejidos y endotelio vascular. Convierte el plasminógeno unido a la fibrina en plasmina |
| Urocinasa | Activa directamente el plasminógeno. No tiene especificidad por la fibrina y no es antigénica |
| Prourocinasa | No es eficaz como activador del plasminógeno. Parece tener mayor especificidad por la fibrina |
| Reteplase (Retavase) | Obtenido por ingeniería genética a partir del tP-A, con mayor potencia trombolítica, mayor vida media. |
| TNK (rtP-A TNK) | Formado por 3 mutaciones del rt-PA con mayor vida media, resistencia al PAI-1, mayor potencia al trombo arterial rico en plaquetas. |
| Estafilocinasa (Star, Sak) | Activador del plasminógeno, fibrina-específico producido por el Staphylococcus aureus. Obtenido por ingeniería genética. |
| Activador del plasminógeno derivado de saliva de vampiro | Clonado de la saliva de algunos vampiros, requiere fibrina como cofactor. |
| Fibrolasa | Actúa rompiendo a la cadena alfa (mayor afinidad) y beta de la fibrina humana, aun en estudio. |
| KiK2Pu | La trombólisis se da en ausencia de la activación del sistema fibrinolítico y sin romper el fibrinógeno. |
ANTIAGREGANTES PLAQUETARIOS
Una de las primeras reacciones después de una lesión endotelial es la adhesión plaquetaria. Esta reacción puede dar por resultado una trombosis. Dentro de los antiagregantes se incluyen los inhibidores de la síntesis de prostaglandinas y del tromboxano A2; el tromboxano A2 es un estimulante muy potente de la agregación plaquetaria y además posee un fuerte efecto vasoconstrictor. El ácido acetilsalicílico posee una acción antitrombótica produciendo acetilación irreversible de la ciclooxigenasa e impidiendo la síntesis de tromboxano A2. El efecto del ácido acetilsalicítico persiste durante la vida de la plaqueta ( 8 a 10 días). La inhibición de la síntesis de tromboxano A2 se puede lograr con dosis muy bajas de ácido acetilsalicílico (40-80 mg/24 h). Con dosis más elevadas se tiene la desventaja de que también se inhibe la ciclooxigenasa de la pared vascular y con ello la síntesis de la prostaciclina, un potente inhibidor de la agregación plaquetaria. Los demás inhibidores de la ciclooxigenasa tiene efectos menos potentes(54). Sustancias que incrementan el AMPc plaquetario, como el dipiridamol, actúan por incremento del AMP cíclico en las plaquetas causando la inhibición de muchas funciones plaquetarias. El AMPc aumenta por estimulación de la adenilciclasa o por inhibición de la fosfodiesterasa. La ticlopidina interactúa con la glucoproteina plaquetaria IIb/IIIa de manera desconocida, para inhibir la unión del fibrinógeno a plaquetas activadas.
La aspirina se ha utilizado para el control y evaluación de otros agentes antiplaquetarios, como la ticlopidina, el clopidogrel y aspirina más dipiridamol de liberación prolongada. La F.D.A (Food and Drug Administration) y el A.C.C.P de los Estados Unidos (American College of Chest Physicians) recientemente recomiendan una dosis de aspirina de 50-325mg/dia para la prevención de ataques isquémicos. Otros agentes antiplaquetarios que han mostrado ser, al menos tan efectivos como la aspirina, son ticlopidina, clopidogrel, y la combinación de bajas dosis de aspirina más dipiridamol de liberación prolongada. Cada uno de estos agentes se recomiendan para prevenir ataques recurrentes. La interpretación de las comparaciones en cuanto a su eficacia, así como las diferencias de sus efectos secundarios, costo y disponibilidad determinará la prescripción para un individuo en particular(59).
INHIBIDORES DE LA TROMBINA
Los inhibidores de la trombina como la hirudina, hiruloq y el argatroban recombinantes se han investigado para diversas indicaciones clínicas. Asímismo, el incremento del interés se debe a la trombocitopenia inducida por la heparina, y la necesidad de efectuar anticoagulación con anticoagulantes alternativos. Estos medicamentos originan una respuesta anticoagulante directa al dirigirse a la trombina(60). La mayoría de estos fármacos (hirudina, refludan, novastan, PEG-hirudina, etc.) se encuentran en proceso de investigación.
TRATAMIENTO TROMBOLITICO
Hasta hace poco tiempo, el sistema plasminógeno-plasmina se contemplaba únicamente como un mecanismo para la fibrinólisis. Hoy en día, una visión más amplia del fibrinógeno permite contemplar su papel como modulador de los procesos hemostático y trombótico en el interior de los vasos. Además de unirse a la fibrina, el plasminógeno también se une a la membrana plaquetaria, a la superficie del endotelio y a la matriz intraplaquetaria que se forma durante la agregación. En cualquier punto donde se fije el plasminógeno, el activador hístico del plasminógeno (t-PA) se fija en las proximidades. El activador también estimula la fibrinólisis y facilita la lisis de los trombos a través de otros mecanismos. Cuando el t-PA se fija a las membranas plaquetarias, la plasmina recién formada adquiere la capacidad para degradarlas glucoproteínas IIb/IIIa, que son necesarias para la adhesión y agregación plaquetaria (61). La activación farmacológica del sistema plasminógeno-plasmina presenta efectos sistémicos. Durante el tratamiento trombolítico sistémico, se perfunde un activador del plasminógeno en la circulación a concentraciones suficientes como para incrementar el nivel circulante del activador en varios cientos o miles por encima del nivel basal. El activador difunde en el coágulo en cantidades suficientes como para activar el plasminógeno unido a fibrina y para acelerar la lisis de un trombo(8, 62).
TRATAMIENTO TROMBOLÍTICO E INFARTO DEL MIOCARDIO
El tratamiento trombolítico parece disminuir el número de muertes por insuficiencia cardiaca, debido principalmente al restablecimiento del flujo sanguíneo miocárdico, a la limitación del tamaño del infarto y a la mejoría de la función ventricular, secundarios a la lisis del trombo intracoronario principalmente. Tras la administración de estreptocinasa, los niveles plasmáticos de fibrinógeno disminuyen en aproximadamente un 70-80%. La consiguiente rápida disminución de la viscosidad sanguínea incrementa el flujo sanguíneo miocárdico en las zonas isquémicas, limitando de esta forma el tamaño del infarto. También permite mantener el flujo sanguíneo en la arteria coronaria recanalizada (disminuyendo el riesgo de una nueva trombosis) y disminuye el trabajo cardíaco. El t-PA que da lugar a menos efectos sistémicos, puede conllevar a un mayor riesgo de retrombosis, de forma que debería administrarse conjuntamente con heparina(63,64). En el infarto de miocardio, la reperfusión con un régimen de terapia fibrinolítica, aspirina y heparina no fraccionada, es limitada por una baja rata de reperfusión, una excesiva rata de reoclusión, una dosis limitada al riesgo de hemorragia intracraneal y una relativa competencia con la angioplastia coronaria directa. Posterior a la reperfusión, ocurre una reoclusión temporal o permanente frecuentemente asociada con una alta mortalidad. En pacientes ancianos la hemorragia intracraneal, como complicación del tratamiento, es frecuente. Finalmente, el riesgo de hemorragia es alto cuando estos pacientes tienen que someterse a procedimientos percutáneos poco tiempo después del tratamiento trombolítico. Por lo cual, dadas estas desventajas, se ha recomendado el uso de la mitad de la dosis de la terapia fibrinolítica convencional combinada con dosis altas de glucoproteína IIb/IIIa inhibidora con Abciximab que ha demostrado excelentes resultados clínicos (61).
En cuanto a la elección del tipo de agente trombolítico y basados en dos grandes estudios clínicos: GISSI-2 e ISIS-3(65,66) que evaluaron no solo las comparaciones entre ambos agentes trombolíticos, sino también la efectividad de usar o no un régimen de heparina subcutánea, se llegó a la conclusión de que ambos agentes trombolíticos, la estreptocinasa y el activador tisular del plasminógeno eran similares en cuanto al efecto trombolítico sin diferencias en cuanto a la mortalidad a los 35 días ni en la sobrevida a los 6 meses de seguimiento, pero el riesgo de hemorragias menores, de hemorragias intracraneales, de alergia y de hipotensión fue importante con ambos agentes trombolíticos, riesgo que se incrementaba con la combinación de éstos con heparina SC (67).
El tratamiento trombolítico es adecuado para la mayoría de los pacientes con infarto de miocardio agudo, con un cuadro de dolor típico cardiaco, persistente, mayor de 15 minutos y menor de 12 horas, (ya que el beneficio del tratamiento es mayor cuando se administra con menos de seis horas de evolución; de seis a doce horas tiene menor beneficio pero es aun de utilidad y en más de doce horas existe poco beneficio aparente cuando el dolor persiste al igual que la elevación del segmento ST) con onda Q en evolución, y con elevación de 1 mm o más del segmento ST en al menos 2 derivaciones de cara inferior, anterior, o lateral, elevación del ST mayor de 2mm en dos o más derivaciones contiguas precordiales, bloqueo reciente de la rama izquierda del haz de Hiss, depresión del ST con R prominente en las derivaciones V2 y V3, si esto hace pensar en un infarto de cara posterior. En cuanto a la edad, en los menores de 75 años se obtiene definitivamente beneficio mientras que en los mayores de 75 años se obtiene poco beneficio, pero la mortalidad aumenta sin la terapia(8,9) .
OTRAS INDICACIONES POSIBLES DEL TRATAMIENTO TROMBOLÍTICO
1. Tromboembolismo venoso y pulmonar: la posibilidad de la lisis de una TVP y embolia pulmonar es mayor tras el tratamiento trombolítico que tras anticoagulación con heparina o warfarina. La dosis recomendada para el tratamiento trombolítico en la embolia pulmonar es de 100 mg de t-PA perfundido durante 2 horas, estreptocinasa bolo endovenoso de 250.000 Ud en 20 minutos, seguido por 100.000 Ud/hora por 24 horas (previamente indicar 100 mg de hidrocortisona EV) o Urocinasa 4.400 Ud/Kg en bolo endovenoso, seguido por una infusión de 4.400 Ud/Kg/hora por 24 horas. Debe reservarse para los pacientes con embolismo severo, alteraciones electrocardiográficas, hipertensión pulmonar, insuficiencia cardiaca derecha o inestabilidad hemodinámica, porque el riesgo de complicaciones hemorrágicas es mucho mayor que con los anticoagulantes. En la TVP, solo se justificaría el tratamiento trombolítico en caso de embolismo masivo que amenaza la supervivencia de la pierna (8).
2. Trombosis arterial periférica: el tratamiento trombolítico perfundido a nivel local demuestra buenos resultados en el 67% de los casos tratados con estreptocinasa, en el 81% de los tratados con urocinasa y en el 90% de los tratados con t-PA con el objetivo de lisar el trombo y restablecer el flujo sanguíneo (8).
3. Angina de pecho inestable: los pacientes con angina inestable presentan con frecuencia trombos coronarios murales no oclusivos situados sobre placas ateromatosas fisuradas y tienen riesgo de oclusión trombótica completa, infarto de miocardio y muerte súbita. Por tanto, es lógico tratar a estos pacientes con tratamiento trombolítico, sin embargo este régimen no ha demostrado mayor efectividad que el tratamiento anticoagulante y antiplaquetario, el cual es suficiente para inhibir el crecimiento de los trombos fibrinoplaquetarios en una arteria coronaria parcialmente ocluida(68).
4. Accidente cerebro-vascular isquémico: el uso de esta terapia se basa en la fisiopatología de los ACV isquémicos, (zona de penumbra) sin embargo en cuanto a la estreptocinasa varios estudios fueron interrumpidos, por lo que su uso no esta recomendado. El t-PA es una terapia de elección en los eventos isquémicos cerebrales, que se acepta aunque exista un riesgo considerable de hemorragias cerebrales y sistemáticas, pero el grado de resolución y permeabilidad es bueno, por lo que se recomienda su uso en manos con mucha experiencia y en centros adecuados(9). Existen estudios con el uso de la urocinasa en las primeras 4 horas del evento en los cuales la recanalización fue significativa, observándose los mejores resultados en el compromiso de las arterias cerebral media y basilar.
5. Derrame pleural paraneumónico complicado: si no hay respuesta en 24 horas con el drenaje cerrado por toracotomía, en vista de la posibilidad de compartamentalización por alta concentración fibrinoide en la cavidad pleural, se pueden usar a través del tubo de tórax estreptocinasa 250.000 UI reteniéndolo por 3 horas durante 3 días, o urocinasa 100.000 UI al día por 3 días, obteniéndose buenos resultados.
| HNF | Tinzaparina (longiparin) | Dalteparina (fragmin) | Enoxapaina (lovenox) | Nadoparina (flaxiparin) | Clivarina (reviparin) | Ardeparina (normoflo) | |
| Peso Molecular Medio | 12000 a 15000 | 5866 | 5819 | 4371 | 4855 | 4653 | 6000 |
| Proporción anti-Xa/anti-Iia | 1:1 | 1,9:1 | 2,1:1 | 2,7:1 | 3,2:1 | 3,6:6.16 | 2:1 |
| Biodisponibilidad a dosis bajas | ‚ | ·90% | ·>90% | ·>90% | ·>90% | ·>90% | ·>90% |
| Depuración dosis dependiente | + | - | - | - | - | - | - |
| Inhibida por factor plaquetario IV | + | +++ | +++ | +++ | +++ | +++ | +++ |
| Inhibe el Xa unido a plaqueta | - | + | + | + | + | + | + |
| Inhibe la función de las plaquetas | ++++ | ++ | ++ | ++ | ++ | ++ | ++ |
| Incrementa la permea-bilidad vascular | + | - | - | - | - | - | - |
| Semidesintegración | ‚ | · | · | · | · | · | · |
Anti-Xa = antifactor (X) diez activado
Anti-IIa = antifactor (II) dos activado
CONTRAINDICACIONES DEL TRATAMIENTO TROMBO-LITICO
Entre las absolutas tenemos: alteración de la conciencia, hemorragia activa, alteraciones de la hemostasia, traumatismo grave reciente, intervención quirúrgica menor a 10 días, intervención invasiva menor a 10 días, intervención neurológica menor de 2 meses, hemorragia gastrointestinal o genitourinaria menor de 10 días, resucitación cardiopulmonar avanzada prolongada mayor de 10 minutos, antecedentes de tumor cerebral, aneurismas, malformaciones de la médula espinal o Malformación Arterio Venosa (MAV), pericarditis aguda, ictus/accidentes isquémicos transitorios menor de doce meses, sospecha de disección aórtica, embarazo, presión arterial persistente >200/120 mmHg, ulcera péptica activa, alergia previa a un producto de estreptocinasa (solo es contraindicación para el uso de estreptocinasa), enfermedad inflamatoria intestinal activa, trauma o cirugía de menos de dos semanas, que podría sangrar hacia un espacio cerrado (69).
Contraindicaciones relativas: trauma mayor o cirugía, mayor de 2 semanas y menor de 2 meses, disfunción hepática significante, presión arterial sistólica >180 mmHg, presión arterial diastólica >110 mmHg, historia de hipertensión arterial crónica no controlada, con o sin tratamiento, endocarditis bacteriana, retinopatía diabética hemorrágica, antecedentes de hemorragia intraocular, ictus/ ataques isquémicos transitorios menor de 12 meses, resucitación cardiopulmonar avanzada breve menor de 10 minutos, tratamiento crónico con warfarina, insuficiencia renal grave, cáncer terminal u otra enfermedad en estadio final, infección estreptocócica reciente, si se va a usar estreptocinasa o anistreptasa, menstruación y post-parto reciente(70).
REFERENCIAS BIBLIOGRAFICAS
1. Baker Jr WF. Outcome analyses for treatment in 100 patients with deep vein thrombosis. Clin Appl Thrombosis/Hemostasis 1.995; 1: 39-48. [ Links ]
2. Hamerschlak N, Rosenfeld L; Utilizaçao da heparina e dos anticoagulantes orais na prevençao e tratamento da trombose venosa profunda e da embolia pulmonar. Aequivos Brasileiros de cardiología 1.996; 67(3):1-9. [ Links ]
3. Anderson FA, Wheeler HB, Goldberg RJ, et al. A population based perspective of the hospital incidence and case fatality rates of deep vein thrombosis and pulmonary embolism. Arch Intern Med 1.991; 151: 933-41. [ Links ]
4. Navarro JL, Avello AG, Pardo A, Cesar JM. Resistencia a la proteína C activada (PCA) crónica de un descobrimiento. Rev Iberoamer Thromb Hemostasia 1.992; 18: 267-73. [ Links ]
5. Tabernero MD, Tomas JF, Alberca I. Incidence and clinical characteristics of hereditary disorder associated with venous thrombosis. Am J Hematol 1.991;36: 249-55. [ Links ]
6. Reimer KA, Lowe JE, Rasmussen MM and Jennings RB. The wavefront phenomenon of ischemic cell death. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1.977; 56(5): 786-794. [ Links ]
7. Ambrose JA, Dangas G. Unstable angina: current concepts of pathogenesis and tratment. Arch Intern Med, 2.000;160(1): 25-37. [ Links ]
8. Claude R, Susan H, Vernon A y James T. Tratamiento trombolítico: revisión de los conocimientos actuales. Hospital Practice 1.992; 7 (10): 57-68. [ Links ]
9. Vásquez Y, Yanez C, Maestre C, Contreras F, Velasco M, Actualización en terapia trombolítica. Arch Vzlanos de Farmac y Terap 1999;18:59-70. [ Links ]
10. Majerus P, Broze G, Tollefsen M. Agentes anticoagulantes, trombolíticos y antiplaquetarios. In: Goodman y Gilman, Las Bases Farmacológicas de la terapéuticas. Editorial McGRAW – HILL INTERAMERICANA; 8vaEd; 1.996. pp. 1273. [ Links ]
11. Lane DA: Heparin binding and neutralizing protein. In Lane DA, Lindahl U (eds): Heparin, Chemical and Biological Properties, Clinical Applications. London, Edward Arnold 1989; 363-391 [ Links ]
12. Backstrom G, Hook M, et al: Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci 1979;76:3198-3206 [ Links ]
13. Lindahl U, Thunberg L, Backstrom G, et al: Extension and structural variability of the antithrombin-binding sequence in heparin. J Biol chem1984; 259:12368-12376. [ Links ]
14. Rosenberg RD, Lam L: Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979;76:1218-1222. [ Links ]
15. Salzman EW, Rosenberg RD, Smith MH, et al: Effects of heparin and heparin fractions on platelet aggregation. J Clin Invest 1980; 64:65-73. [ Links ]
16. Graham FP, Russell DH: Heparina no fraccionada y de bajo peso molecular : Comparaciones y recomendaciones actuales. Clin Med NorAm 1998; 3:553-564 [ Links ]
17. Barzu T, Molho P, Tobelem G, et al: Binding of heparin and low molecular weight heparin fragments to human vascular endothelial cells in culture. Nouv Rev Haematol 1984;26:243-247 [ Links ]
18. Bjornsson TO, Wolfram BS, Kitchell BB: Heparin kinetics determined by three assay methods.Clin Pharmacol Ther 1982;31:104-113 [ Links ]
19. Hoppensteadt D, Walenga JM, Fasanella A, et al: TFPI antigen levels in normal human volunteers after intravenous and subcutaneous administration of unfractionated heparin and low molecular weight heparin. Thromb Res 1995; 77:175-185. [ Links ]
20. Gallus AS, Hirsh J, Tuttle RS el al.Small Subcutaneous doses of heparin in prevention of venous thrombosis. N Engl J Med 1.973; 288: 545-52. [ Links ]
21. Kakkar VV, Corrigan T, Spindler J et al. Efficacy of low doses of heparin in prevention of deep vein thrombosis after major surgery. A doble blind, randomised trial. Lancet 1.972; 2: 101-106. [ Links ]
22. Brill-Edwards P, Ginsberg S, Johnston M, et al: Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119:104-109 [ Links ]
23. Brandjes DPM, Heijboer H, Buller HR,et al: Acenocoumarol and heparin compared with acenocoumarol alone in the initial treatment of proximal-vein thrombosis. N Engl J Med 1992; 327:1485-1489 [ Links ]
24. Hull RD, Raskob GE, Hirsh J, et al: Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal- vein thrombosis. N Engl J Med 1986; 315:1109-1114. [ Links ]
25. Raschke RA, Reilly BM, Guidry JR, et al: The weight- based heparin dosingnomogram compared with a " standard care" nomogram. Ann Intern Med 1993; 119:874-881 [ Links ]
26. Boshkov LK, Warkentin TE, Hayward CPM, et al: Heparin-induced thrombocytopenia and thrombosis: Clinical and laboratory studies. Br J Haematol 1993;84:322-328 [ Links ]
27. Kelton JG, Sheridan D, Santos A, et al: Heparin-induced thrombocytopenia: Laboratory studies. Blood 1988; 721:925-930. [ Links ]
28. Warkentin TE, Kelton JG: Heparin-induced thrombocytopenia. Prog Hemost Thromb 1991; 10:1-34 [ Links ]
29. Hirsh J, Raschke R, Warkentin TE, et al:Heparin Mechanism of action, pharmacokinetics, dosing considerations, monitoring, efficacy, and safety. Chest1995; 108:258S-275S. [ Links ]
30. Barrowcliffe TW. Low molecular weight heparin(s). Br J Haematol 1.995; 90: 1-7. [ Links ]
31. Thomas DP. Does low molecular weight heparin cause less bleeding? Thromb Haemost 1.997; 78: 1422-5. [ Links ]
32. Hirsh J, Levine MN. Low molecular weight heparin. Blood 1.992; 79: 1-17. [ Links ]
33. Nurmohamed MI, Rosendaal FR, Buller HR: LMWH vs standard heparin for the prevention of venous thrombo-embolism in general and orthopedic surgery. A metaanalysis. Lancet 1.992; 340: 152-6. [ Links ]
34. Leizorovicz A, Haugh MC, Clapuis FR, et al. Low molecular weight heparin in prevention of perioperative thrombosis. Br Med J 1.992; 305: 913-20. [ Links ]
35. Prandani P, Mannucci PM. Deep vein thrombosis of the lower limbs: Diagnosis and management. Thromb Hemost 1.994; 7: 693-712. [ Links ]
36. Leclerc JR, Geets WH, Desjardins L, et al. Prevention of venous thromboembolism after knee arthroplasty. A randomized, double blin trial comparing enoxaparin with warfarin. Ann Intern Med 1.996; 124: 619-26. [ Links ]
37. Hirsh J. The emerging role of low-molecular-weight heparin in cardiovascular medicine. Prog Cardiovasc Dis, 2.000; 42 (4): 235-246. [ Links ]
38. Wallentin L. Efficacy of low-molecular-weight heparin in acute coronary syndromes. Am Heart J 2.000; 139(2): 29-32. [ Links ]
39. Anderson L-O, Barrowcliffe TW, Holmer E, et al: Molecular weight dependency of the heparin potentiated inhibition of thrombin and activated factor X : Effect of heparin neutralization in plasma. Thromb Res 1979;115:531-538 [ Links ]
40. Bara L, Samana MM: Pharmacokinetics of low molecular weight heparins. Acta Chir Scand 1988; 543:65-72. [ Links ]
41. Briant L, Caranobe C, Saivin S, et al: Unfractionated heparin and CY216: Pharmacokinetics bioavailabilities of the anti-factor Xa and IIa: Effects of intravenous and subcutaneous injection in rabbits. Thromb Haemost 1989; 61:348-353 [ Links ]
42. Carter CJ, Kelton JG, Hirsh J, et al: The relationship between thr hemorrhagic and antithrombotic properties of low molecular weight heparins and heparin. Blood 1982; 59:1239-1245 [ Links ]
43. Fareed J, Walenga JM, Hoppensteadt D, et al: Comparative study on the in vitro and in vivo activities of seven low-molecular weight heparins. Haemostasis 1988; 18:3-15. [ Links ]
44. Fareed J, Walenga JM, Racanelli A, et al: Validity of the newly established low molecular weight heparin standard in cross referencing low molecular weight heparins. Haemostasis 1988;3:33-47. [ Links ]
45. Holmer E, Soderberg k, Bergqvist D, et al: Heparin and its low molecular weight derivatives: Anticoagulant and antithrombotic properties. Haemostasis 1986; 16: 1-7 [ Links ]
46. Andrew M, Cade J, Buchanan MR, et al: Low-molecular weight heparin does not cross the placenta. Thromb Haemost 1983;50:225. [ Links ]
47. Forestier F, Daffos F, Capella-Pavlovsky M: Low molecular weight heparin (PH 10169) does not cross the placenta during the second trimester of pregnancy: Study by direct fetal blood sampling under ultrasound. Thromb Res 1984;34:557-560. [ Links ]
48. Ginsberg JS, Hirsh J: Use of antithrombotic agents during pregnancy. Chest 1995;108:305S-311S. [ Links ]
49. Schofield FW: Damage sweet clover: The cause of the new disease in cattle simulating hemorrhagic septicemia and black leg. J Am Vet Assoc 1924; 64:553-575 [ Links ]
50. Roderick LM: Problems in the coagulation of the blood. Am J Physiol 1931;96:413-425 [ Links ]
51. Quick AJ : The prothombin time in haemophilia and in obstructive jaundice. J Biol Chem1935; 109:73-74 [ Links ]
52. Link KP: The anticoagulant from spoiled sweet clover hay. Harvey Lect 1994; 34:162-216. [ Links ]
53. Link KP: The discovery of dicumarol and its sequels. Circulation 1959;19:97-107. [ Links ]
54. Giuliani R, Szwarcer E, Bendetowicz AV. Heparina y heparinoides. In: Kordich LC, Avalos JCS, Vidal HO, Guerra CCC- Manual de hemostasia y trombosis. "nd ed. Grupo CLAHT, 1.990;1: 82-86. [ Links ]
55. Brigden ML, Oral anticoagulant therapy. Postgrad Med 1996;99:81-102. [ Links ]
56. Gladman JR, Dolan G : Effect of age upon the induction and maintenance of anticoagulation with warfarin. Postgrad Med J 1995;7:153-155. [ Links ]
57. Kumar S, Haigh JR, Rhodes LE, et al: Poor compliance is a major factor in unstable outpatient control of anticoagulant therapy. Thromb Haemost 1989;62:729-732. [ Links ]
58. Brien WF, Crawford L, Wood DE: Discrepant results in INR testing. Thromb Haemost 1994;72:985-989 [ Links ]
59. Gregory WA, Jan GP, Antiplatelet therapy: New foundations for optimal treatment decisions. Neurology 1999;53:S25-S31 [ Links ]
60. Fareed J, Callas Demetra, Hoppensteadt DA, et al: Fármacos antitrombina como anticoagulantes y antitrombóticos: Inferencias en la creación de fármacos. Clin Med NorAm 1998;3:535-551 [ Links ]
61. Califf RM. Combination therapy for acute myocardial infarction: Fibrinolytic therapy and glycoprotein Iib/IIIa inhibition. Am Heart J 2000;139: (2) 33-37. [ Links ]
62. Laffel GL, Braunwald E. Thrombolytic therapy: A new strategy for treatment of acute myocardial infarction. N Engl J Med 1.984; 311: 710. [ Links ]
63. TIMI study group. The thrombolysis in myocardial infarction (TIMI) trial. Phase I findings. N Engl J Med 1.985; 312:932.24.- [ Links ]
64. Yusuf SW et al. Intravenous and intracoronary fibrinolytic therapy in acute myocardial infarction: Overview of results on mortality, reinfarction and side effects from 33 randomized control trials. Eur Heart J 1985; 6: 556-566. [ Links ]
65. The International study group. In Hospital motality and clinical course of 20.891 patients with suspected acute myocardial infarction randomised between alteplase and streptokinase with or without heparin. The Lancet 1990; 336: 71-75. [ Links ]
66. ISIS-3: A randomised comparison of streptokinase vs tisse-type plasminogen activator vs anistreplase and of aspirin plus heparin vs aspirin along among 41.299 cases of suspected acute myocardial infarction. The Lancet 1992; 339:753-770. [ Links ]
67. Bates ER and Topol EJ. Limitation of thrombolytic therapy for acute myocardial infarction complicated by congestive heart failure and cardiogenic shock. J Am Coll Cardiol 1.991; 18(4): 1.077-1.084. [ Links ]
68. Sobel BE. Coronary Thrombolysis and the new biology. J Am Coll Cardiol 1.989; 14: 850. [ Links ]
69. Tollefsen DM, Majerus DW, Blank MK: Heparin cofactor II: Purification and properties of heparin- dependent inhibitor of thrombin in human plasma. J Biol Chem1982; 257:2162-2169. [ Links ]
70. Belsey RE, Fischer PM, Baer DM: An evaluation of a whole blood prothrombin analyzer designed for use by individuals without formal trainig. J Fam Pract1991; 33:266-271. [ Links ]
