








Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
Print version ISSN 0798-0264
AVFT vol.20 no.1 Caracas Feb. 2001
Archivos Venezolanos de Farmacología y Terapéutica, Volumen 20 - Número 1, 2001 (43-51)
Utilidad y ventajas del uso de Modelos Matemáticos en el estudio de la Insulinoresistencia y función de la Célula Beta Pancreática. Homeostasis Model Assesstment (Homa): Fundamento y bases para su implementación en la práctica clínica
V Bermúdez1, C Cano1, M Medina1 y M Núñez1.
- Centro de Investigaciones Endocrino-Metabólicas "Dr. Félix Gómez". Facultad de Medicina. La Universidad del Zulia.
RESUMEN
La Diabetes mellitus es un síndrome metabólico que se caracteriza por la presencia de hiperglucemia debido a una deficiencia en la secreción de insulina o a una combinación de insulino-resistencia (IR) con inadecuada secreción de insulina. La diabetes tipo 2 constituye la forma más prevalente y resulta de IR, causada principalmente por obesidad central con un defecto en la secreción de insulina. Han sido desarrollados varios métodos experimentales que tratan de determinar la IR en humanos. La técnica del clamp euglicémico-hiperinsulinémico es el método "gold standard" para su cuantificación in vivo, sin embargo, no puede ser fácilmente aplicado a estudios de grandes poblaciones ya que necesita una infusión endovenosa de insulina, múltiples tomas de muestras sanguíneas durante 3 horas y un continuo ajuste de una infusión de glucosa para cada individuo. El Homeostasis model assessment (HOMA) constituye uno de los métodos más simples que provee índices de insulino-resistencia (HOMA–IR) y funcionalismo de la célula beta (HOMA
b-cell) con sólo la determinación de concentraciones basales de glucosa e insulina plasmáticas, lo que la hace aplicable a estudio de gran escala. El HOMA no sólo se limita a pacientes diabéticos, sino que también puede ser aplicado a individuos cuya patología involucre IR o alteración del funcionalismo de la célula beta.Palabras Claves: Diabetes mellitus, Insulino resistencia, Clamp euglicémico - hiperinsulinémico, HOMA.
ABSTRACT
Diabetic mellitus is a syndrome with disordered metabolism and inappropriate hyperglycemia due to either a deficiency of insulin secretion or to a combination of insulin resistance and inadequate insulin secretion to compensate. Type 2 diabetes is the most prevalent form and results from insulin resistance, mainly caused by visceral obesity, with a defect incompensatory insulin secretion. Various experimental methods and modelling approaches have been developed to assess in vivo insulin resistance in men, the hyperinsulinemic euglicemic glucose clamp technique is the "gold standard" for quantifying sensitivity in vivo, however, the glucose clamp is no easily applied in large scale investigations because IV infusion of insulin, frequent blood samples over a 3-h period, and continuous adjustment of a glucose infusion are required for each subjects studied. Homeostasis model assessment (HOMA) approach is one of the simplest methods that provide indices of insulin secretion and insulin resistance derived from fasting plasma glucose and fasting plasma insulin levels, thus, it can be reliably used in large – scale or epidemiological studies in which only a fasting blood sample is available to assess insulin sensitivity. Therefore, the ability to easily quantify insulin sensitivity in large number of subjects may be useful for investigating the role of insulin resistance in the pathophysiology of these major public health problems.
Key Words: Mellitus diabetic, Resistance insulin, Hyperinsulinemic euglicemic clamp, Homa.
INTRODUCCIÓN
La Diabetes Mellitus tipo 2 es la endocrinopatía que aqueja con mayor frecuencia al ser humano(1). Se caracteriza fundamentalmente por la presencia de insulinoresistencia (IR) y/o por defectos en la secreción de insulina. Una gran cantidad de estudios han comprobado de forma inequívoca que la IR y la hiperinsulinemia son predictores muy confiables para el desarrollo de diabetes tipo 2(2,3). Igualmente, otras líneas de investigación sugieren que una secreción anormal de insulina (evaluada por la respuesta aguda de secreción de insulina a glucosa endovenosa) predice igualmente el desarrollo de diabetes tipo 2, especialmente en aquellos individuos con intolerancia a la glucosa(2,4).
A lo largo del tiempo el clamp hiperinsulinémico-euglicémico, el test de supresión de secreción de insulina y el test de tolerancia intravenosa a la glucosa con toma de múltiples muestras y modelado por computadora (FSIGT) han sido utilizados para la evaluación de la IR siendo todos complicados, costosos y de muy difícil aplicación en grandes poblaciones(5,6,7). Además de lo antes planteado, solo el FSIGT (a través del cual se calcula la respuesta insulínica aguda) puede utilizarse para estimar la capacidad insulinosecretora pancreática.
Debido a los inconvenientes presentados por los métodos precitados, se ha creado un enorme interés sobre la posibilidad del desarrollo de modelos matemáticos, que expresen las alteraciones metabólicas involucradas en la fisiopatología de enfermedades humanas, entre ellas, la misma diabetes Mellitus(8,9,10,11).
La dinámica de la interacción entre la masa de células beta funcionales, la capacidad de secreción de insulina, la concentración plasmática de glucosa, la resistencia tisular periférica (tejido muscular y adiposo) y la resistencia a la insulina por parte del hígado son los principales elementos utilizados en la generación de simulaciones sobre la progresión de la diabetes mellitus tipo 2(11-13).
El Homeostasis Model Assessment u HOMA, es un modelo matemático desarrollado hace más de quince años por DR Matthews, RC Turner y colaboradores del Laboratorio de Investigación de Diabetes de Radcliffe, Oxford UK, como una forma alternativa de cuantificar la IR y disfunción de la célula beta en el ser humano con sólo dos variables de muy fácil cuantificación: La glucosa e insulina plasmática en ayuno(9,12).
UNA VISIÓN GLOBAL DE LA HOMEOSTASIS DE LA GLUCOSA PLASMÁTICA
El papel central de la insulina en el control de la concentración plasmática de la glucosa puede dividirse de manera conceptual en el efecto recíproco entre, su secreción y la respuesta o sensibilidad de los tejidos a ésta(14). Poco después del descubrimiento de la insulina, Himsworth puso en evidencia que la diabetes podía dividirse en dos grupos: uno, sensible a la insulina y otro insensible a la misma(15). Solo hubo que esperar pocos años para que el desarrollo de técnicas radioinmunológicas comprobaran que era cierto que existían diferentes grados de sensibilidad a la insulina (y concentraciones plasmáticas de glucosa) en un grupo poblacional determinado con un buen estado de salud, representando en realidad, un espectro continuo donde la resistencia a la insulina observada en la obesidad, intolerancia a la glucosa y diabetes se ubican en su extremo patológico(16).
No se debe olvidar que la disminución de la capacidad de respuesta de los tejidos a la insulina puede ser causada por una gran variedad de condiciones como la inactividad física, alteraciones en el estado general, infecciones, patologías hormonales (acromegalia, síndrome de Cushing, glucagonoma, etc.) y la hiperglucemia crónica misma observada en la diabetes mal controlada(17).
La célula beta no debe considerarse como una inyectadora endógena de insulina; ciertamente, el diseño estructural y su ubicación son un privilegio de millones de años de evolución. Para ilustrar un poco este hecho, basta recordar que la insulina secretada por la célula beta se dirige directamente a la circulación portal y de allí al hígado, donde es capaz de bloquear la gluconeogénesis y estimular la glucogenosíntesis, la glucólisis y la vía de las pentosas, entre otras. La insulina que se suministra a los pacientes bien sea por vía subcutánea o por vía endovenosa jamás alcanza la elevada concentración observada en el sistema portal, motivo por el cual muchas veces no se evita de manera completa la gluconeogénesis. En la secreción de insulina están involucrados complejos mecanismos que aseguran su liberación en forma directamente proporcional con la concentración de glucosa.
Bases moleculares de la secreción de insulina
La célula beta como cualquier célula endocrina pertenece al grupo de las células excitables. Esto quiere decir que tiene una maquinaria proteica ejemplar en forma de canales iónicos, bombas, receptores de membranas y proteínas reguladoras que trabajan al unísono con la finalidad de cambiar el potencial eléctrico transmembrana según nos encontremos en ayuno (inhibición de la secreción de insulina) o en el período post-prandial inmediato (estimulación de la secreción de insulina).
Lo primero que debe considerarse en la célula b es la presencia de canales iónicos como el canal de goteo de potasio y los canales de calcio voltaje dependientes(18,19). Cuando la célula beta se encuentra en reposo durante el ayuno (secreción mínima de insulina) los canales de goteo de K+ se encuentran abiertos. Estos canales son regulados por el voltaje intracelular y por el ATP, por lo cual también se les denomina canales de K+ dependiente del ATP (KATP). El canal de KATP sensible es una gran proteína octamérica que se puede dividir en dos partes desde un punto de vista funcional: una, denominada poro, que representa el canal en sí y que recibe el nombre técnico de Kir6.2 y otra, que por su alta afinidad a las sulfonilureas ha recibido el nombre de SUR-1 (Sulfonilurea receptor-1). La existencia de la SUR-1 sugirió por mucho tiempo que debía existir un ligando endógeno para ésta. A finales de la década de los 80, se descubrió el ligando endógeno del receptor para sulfonilureas denominado actualmente a-Endosulfina. Recientemente, su gen fue localizado cromosómicamente, aislado y caracterizado de manera completa(20).
El KATP se encuentra abierto cuando el potencial eléctrico transmembrana esta alrededor de – 65mV. La función principal de este canal es la de permitir, durante el período de reposo, la salida lenta de K+ hacia el espacio extracelular a favor de su gradiente de concentración. Esta fuga lenta de potasio produce pérdida de cargas positivas en el interior celular quedando éste cargado negativamente con respecto al exterior. Durante el período post-prandial, la entrada masiva de glucosa al interior de la célula beta produce un incremento de la concentración de ATP gracias al metabolismo de la glucosa por vía glucolítica. El ATP rápidamente se une al canal KATP cerrándolo debido a la actividad reguladora de la
a-Endosulfina lo que ocasiona una acumulación de K+ dentro de la célula. La bomba de Sodio y Potasio (Na+/K+ ATPasa) funciona normalmente por lo que el K+ sigue entrando a la célula. Las cargas positivas aportadas por la acumulación del K+ cambian súbitamente el potencial transmembrana haciéndolo más positivo en el interior (alrededor de –30 mV), esto produce la apertura de los canales de Ca++ voltaje dependiente permitiendo su paso al interior de la célula lo que promueve la contracción microtubular responsable de la migración, contracción y fusión de las vesículas secretoras de insulina con la membrana plasmática(21,22,23).Si hacemos un análisis de este sistema, podemos proponer que pueden presentarse situaciones capaces de interferir con la secreción normal de insulina como:
a) Defectos genéticos o adquiridos (glicosilación, oxidación, etc.) a nivel de los canales de KATP sensibles en la célula
b) Defectos genéticos o adquiridos (bloqueo por antagonistas) de los canales de Ca++ voltaje dependientes en la célula
b.c) Defectos genéticos o adquiridos (glicosilación, oxidación, etc.) de los transportadores de membrana para la glucosa en la célula beta isoforma 2 (GLUT´s 2).
d) Defectos genéticos o adquiridos (glicosilación, oxidación, etc.) en la
a-Endosulfina.e) Defectos genéticos (Glucocinasa) o adquiridos (glicosilación, oxidación, etc.) en la glucólisis de la célula
b.f) Defectos genéticos o adquiridos en las vías metabólicas de la oxidación de ácidos grasos en la célula
b.Bases moleculares de los efectos tisulares de la insulina
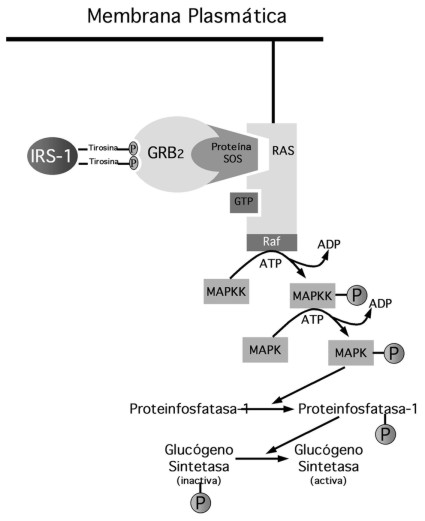
Durante mucho tiempo se especuló sobre los mecanismos moleculares involucrados en la traslocación de la señal insulínica en especies superiores, en especial, en lo que respecta a la generación de los segundos mensajeros de la insulina. Muchas sustancias fueron postuladas como posibles segundos mensajeros para esta hormona, tales como el Calcio, el AMPc, el GMPc, fosfoinositósidos e incluso, hasta el peróxido de hidrógeno. Sin embargo, no fue hasta 1986 cuando Morris White y su grupo de investigación en los EE.UU., descubren el primer segundo mensajero de la insulina, el IRS-1 (Insulin receptor sustrate–1)(24). Aunque el objetivo principal de este artículo no es el de hacer una revisión del mecanismo de acción de la insulina, es importante señalar que desde un punto de vista didáctico se puede dividir en tres grandes eventos. En primer lugar, la estimulación del receptor de insulina mediante la activación de su capacidad intrínseca enzimática tipo tirosincinasa y la fosforilación del IRS-1 (Figura 1). En segundo lugar, la formación del complejo IRS-1-GRB2-SOS y con esto la activación de la proteína Ras (Figura 1) y en tercer lugar la activación por parte de Ras de una serie de enzimas tipo cinasas (Treonin-Serincinasas) en forma de cascada que finalmente activan las enzimas reguladoras clave de las vías metabólicas influenciadas por la insulina (Figura 2). Teóricamente este sistema puede sufrir, alteraciones innatas o adquiridas que pueden afectar de manera directa la sensibilidad tisular a la insulina(24). De esta forma, resulta obvio que la disfunción innata o adquirida en el mecanismo de síntesis, secreción y transporte plasmático de la insulina, así como de algún trastorno en los mecanismos involucrados en la captación y generación de la señal insulínica pueden desencadenar en una desregulación metabólica de suficiente cuantía que pueda expresarse a través de hiperglucemia y por lo tanto diabetes mellitus. Por este motivo, vemos como imperativa la necesidad de estudiar con profundidad, este sistema en nuestros pacientes y no como una necesidad cosmética o como algo superfluo que le añade costos al manejo y control del paciente aquejado por esta condición. De hecho, cualquier herramienta que pueda estar a nuestra disposición capaz de abrir una luz en la oscuridad de lo que es una entidad verdaderamente compleja en su análisis global debe ser bienvenida aunque sometida al escrutinio del método científico y no excluir su utilización solo por motivos de tipo viscerales, lo cual resulta en extremo dogmático y que no genera en ningún momento beneficios al quehacer científico.
Figura 1: La estimulación del receptor de insulina mediante la activación de su capacidad enzimática tipo tirosíncinasa y la fosforilación del IRS-1 conduce a la formación del complejo IRS-1 – GRB2-SOS, y por ende a la activación de la proteína RAS.
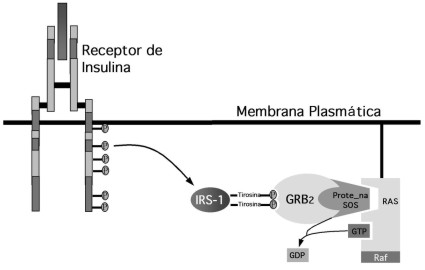
Figura 2: Activación por parte de la proteína RAS de la cascada de fosforilación enzimática tipo cinasas que activan las enzimas reguladoras de las vías metabólicas influenciadas por la Insulina.
EL MODELO MATEMÁTICO HOMA
Como respuesta a los problemas de evaluación de la insulinosensibilidad mediante métodos que dependen de la infusión continua de glucosa y las dificultades experimentadas en la interpretación de pruebas realizadas en estados no estacionarios se han ideado pruebas basadas en modelos matemáticos para estimar la IR y capacidad de secreción de la célula beta(12,13).
La concentración plasmática de la glucosa y de insulina en condiciones de ayuno tanto en individuos sanos como en pacientes diabéticos tipo 2 se ubica en un nivel característico para cada persona y para cada estado nutricional individual. La concentración basal de insulina es consecuencia de la concentración de glucosa en ayuno, de la capacidad secretoria de la célula beta y el ritmo de secreción pulsátil de la insulina (pulsos con aproximadamente 15 minutos de intervalo)(28,29). La hiperglucemia basal que se observa en la diabetes tipo 2 se debe fundamentalmente a una alteración en el sistema de retroalimentación existente entre el hígado y la célula beta, en el cual, esta última es incapaz de controlar la gluconeogénesis hepática y en mucho menor grado la IR periférica(30,31).
El HOMA representa un modelo matemático "estructural" mas que "mínimo" de las interacciones entre la glucosa y la insulina, el cual se ha usado para calcular las diferentes combinaciones de hiperglucemia versus insulina plasmática normal, baja o elevada(32,33). El HOMA incorpora funciones matemáticas separadas que describen las respuestas fisiológicas a la insulina y a la glucosa de los diferentes órganos implicados en la homeostasis de la glucosa tales como el músculo, hígado, tejido adiposo, páncreas, tejidos periféricos relativamente insensibles a la insulina para el transporte de glucosa e incluso, la concentración de péptido C, pro-insulina y hasta las pérdidas renales de glucosa cuando sobrepasan su umbral de reabsorción(34,35). Las predicciones generadas por este modelo están acordes con los resultados obtenidos en seres humanos a través del clamp euglicémico-hiperinsulinémico. De esta forma, se ha creado una prueba formal para determinar el grado de sensibilidad periférica a la insulina y la capacidad de funcionalismo de la célula beta pancreática mediante la determinación de la concentración de glucosa e insulina plasmática en ayuno(32).
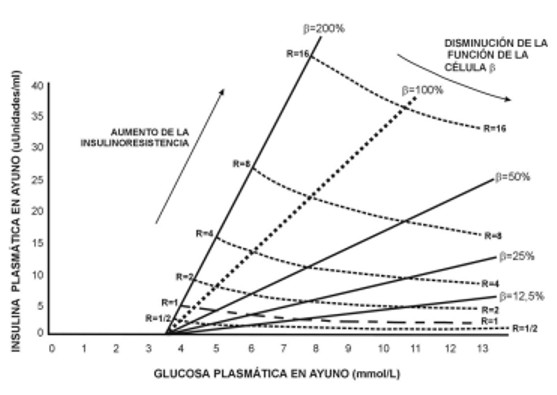
El HOMA en realidad es un modelo generado directamente por computadoras donde al analizar los diferentes elementos que intervienen en la homeostasis de la glucosa (y que a la vez influyen en la secreción de insulina) se genera un gráfico donde el eje de las Y esta representado por la concentración de insulina plasmática en ayuno contra la concentración de glucosa en ayuno (representada en el eje de las X) que se esperarían obtener en los diferentes grados de deficiencia secretora de la célula beta y de insulinoresistencia(36). De esta manera se puede estimar, gracias a este gráfico, la IR y la función de la célula beta que se debería esperar para cualquier paciente diabético conociendo la glucemia e insulina en ayuno.
Los resultados estimados a través del HOMA se han comparado con el Clamp hiperinsulinémico-hiperglucémico(37,38) y el test de tolerancia intravenosa(6) a la glucosa en lo que respecta al funcionalismo de la célula Beta. En cuanto a la insulinoresistencia, se ha comparado con el clamp hiperinsulinémico-euglicémico, el nivel de obesidad, los niveles de insulina en ayuno y estudios celulares en monocitos y eritrocitos. En ambos casos, los coeficientes de correlación han arrojado resultados óptimos en cuanto al desempeño de este modelo en la cuantificación de la IR y un poco menos en la capacidad secretoria de la célula beta pancreática(6,39).
Generación del modelo
La curva de la función de la célula beta se encuentra ubicada muy cerca del estado estacionario de la respuesta del péptido C durante el clamp hiperglucémico a las 2,5 horas practicado con concentraciones de glucosa de 7,5; 10 y 15 mmol/l tal como lo demostró el grupo de Turner en seis sujetos normales y con edades comprendidas entre 40-68 años(12). Estos individuos tuvieron una curva dosis respuesta glucosa: péptido C casi lineal con una intercepción en el eje de las X (concentración de glucosa) de 3,5 mmol/l. Al practicar el clamp a 5 pacientes diabéticos tipo 2 con glucemias en ayuno entre 5,5 a 6,5 mmol/l tuvieron una intercepción en el eje de las X similar a los anteriores con una curva también casi lineal pero con la diferencia que dicha curva estaba desviada a la derecha. La respuesta Glucosa: insulina para los sujetos normales fue: Insulina = 5 (glucosa en ayuno [mmol/l] – 3,5). De esta forma, el modelo permite la determinación de la concentración basal estacionaria de glucosa e insulina que se observa en los diferentes grados de disfunción de la célula beta y de IR. Cualquier combinación de ambas esta emparejada con un único y particular juego de insulinemia y glucemia basal que puede llevarse- a la gráfica generada por el modelo HOMA (Fig. 3) o se puede calcular de manera directa a través de arreglos numéricos con las siguientes fórmulas(34):
Figura 3: Representación gráfica del Homeostasis Model Assesstment (HOMA)
HOMAbetacell = 20 x Insulinaayuno (µUI/ml) / Glucosaayuno (mmol/l) – 3,5
HOMAIR = Insulinaayuno (µUI/ml) x Glucosaayuno (mmol/l) / 22,5
Asumiendo que un individuo es completamente sano, con un índice de masa corporal normal y sin antecedentes familiares de diabetes mellitus se presume que el HOMA beta cell (o funcionamiento de la célula beta calculado por el HOMA) estará alrededor del 100% y el HOMA IR o índice de insulinoresistencia estará muy cercano a 1. Valores por encima de 1 representarán un nivel creciente de insulinoresistencia.
Estudio general de las fórmulas generadas por el modelo matemático HOMA
Obviamente no está a nuestra disposición los datos relacionados directamente con los arreglos numéricos y matemáticos con los que se obtuvieron las fórmulas. La concentración de insulina y glucosa en ayuno y que en realidad representan las variables de entrada (in) con las que queremos obtener un valor de salida (out) representados por la insulinoresistencia y el funcionalismo de la célula beta. El resto de elementos numéricos observados en la fórmula de HOMABcell el 3,5 y el 20 y para la fórmula de HOMAIR el número 22,5. La pregunta pertinente en estos momentos resulta clara ¿de dónde provienen estos números?. La respuesta es muy sencilla, los desarrolló el modelo matemático mismo y a pesar de no tener la data original en nuestras manos, definitivamente sí podemos comprobar la validez de estas constantes fácilmente.
Breve estudio de la fórmula HOMABcell
Si nos concentramos un poco en la fórmula del HOMABcell y
recordamos que un individuo teórico perfecto en cuanto a su capacidad secretora de la célula
b debe tener el HOMABcell con un valor igual o muy aproximado al 100% podemos sustituir este resultado en el lado izquierdo de la expresión anterior, lo cual observamos en la segunda fórmula.
HOMAbetacell = 20 x Insulinaayuno (µUI/ml) / Glucosaayuno (mmol/l) – 3,5
100 = 20 x Insulinaayuno (µUI/ml) / Glucosaayuno (mmol/l) – 3,5
Si analizamos ahora la gráfica generada por el modelo matemático y buscamos en ella una concentración de glucosa en ayuno y la concentración equivalente de insulina plasmática en ayuno que corresponda a valores de funcionalismo de la célula
b del 100%, como por ejemplo glucosa de 4,5 mmol/l e insulina de 5 µUI/ml pueden ser sustituidas en la fórmula anterior:
100 = 20 x 5 (µUI/ml) / 4,5 (mmol/l) – 3,5
Ahora solo queda despejar el número 20 de la fórmula. Para hacerlo de una manera fácil, resolvemos primero la resta en el denominador, es decir, 4,5 – 3,5, lo cual es 1:
100 = 20 x 5 (µUI/ml) / 1
100 x 1 = 20 x 5 (µUI/ml)
O lo que es lo mismo:
100 = 20 x 5 (µUI/ml)
Ahora podemos despejar directamente el número 20 llevando las 5 µUI/ml al otro lado de la expresión (dividiéndola):
100/5=20
Comprobando finalmente que la ecuación es correcta al resolver la división y comprobar que dicha constante se cumple para la pareja glucosa/insulina que habíamos escogido a partir del gráfico:
20=20
Breve estudio de la fórmula HOMAIR
Igualmente que en el caso anterior se puede aplicar el mismo razonamiento para la fórmula que representa la sensibilidad insulínica u HOMAIR:
HOMAIR = Insulinaayuno (µUI/ml) x Glucosaayuno(mmol/l) / 22,5
Para este caso, el valor ideal de sensibilidad insulínica para un individuo es de 1, el cual puede ser sustituido en el miembro situado del lado izquierdo de la fórmula:
1 = Insulinaayuno (µUI/ml) x Glucosaayuno(mmol/l) / 22,5
Podemos utilizar la misma concentración de glucosa con su correspondiente concentración de insulina de la fórmula anterior, es decir, glucosa de 4,5 mmol/l e insulina de 5 µUI/ml y sustituir los valores en la fórmula:
1 = 5 (µUI/ml) x 4(mmol/l) / 22,5
Resolvemos la multiplicación a nivel del numerador:
1 = 22,5 / 22,5
y luego despejamos la constante 22,5 del denominador:
22,5 = 22,5
Comprobamos que la fórmula es correcta al resolver la división y corroborar que dicha constante se cumple para la pareja glucosa/insulina que se había seleccionado.
ALGUNOS ESTUDIOS EPIDEMIOLÓGICOS Y CLÍNICOS QUE AVALAN LA APLICABILIDAD DEL HOMA
La estimación de la insulinoresistencia y de la función de la célula
b mediante el HOMA se adapta perfectamente a estudios epidemiológicos a gran escala. Dos ejemplos recientes son el Estudio de Diabetes de Ciudad de México y El estudio Familiar de Diabetes de Oxford y Northhamptom.El Estudio de Ciudad de México
Haffner y cols. estudiaron el deterioro progresivo de la función de la célula
b y el incremento de la insulinoresistencia en individuos de Ciudad de México con alto riesgo de desarrollar diabetes a través de HOMA durante un periodo de 3,5 años(40). Al culminar el estudio se organizaron dos subgrupos: el primero, conformado por los individuos que no desarrollaron diabetes y el segundo, se conformó con los individuos que desarrollaron diabetes durante el período de estudio. La insulinoresistencia estimada por el HOMA fue invariablemente predictiva para el desarrollo de diabetes mellitus tipo 2, inclusive con un mejor valor predictivo que la insulina en ayuno aislada. A pesar de que la función de la célula b estimada por HOMA no fue predictiva para el desarrollo de diabetes cuando se incluía en el modelo correlacionada junto con la insulinoresistencia, se observó que estaba significativamente asociada de manera inversa al desarrollo de la diabetes, lo que implica que al examinar al individuo durante cierto período de tiempo (de duración variable y particular para cada quién) antes de presentar diabetes establecida, se presenta hiperinsulinemia que en presencia de insulinoresistencia es muy sugestiva del desarrollo posterior de diabetes tipo 2.El Modelo HOMA en El Estudio San Antonio, Texas
El Estudio San Antonio Heart, es una investigación poblacional sobre diabetes y enfermedad cardiovascular realizado en Norteamericanos de origen Mexicano y blancos no hispánicos en los Estados Unidos de América(41). La primera fase de este estudio se desarrolló desde 1979 hasta 1982 y la segunda desde 1984 hasta 1988, completándose una muestra de 2.465 pacientes estudiados (hombres y mujeres no grávidas) entre los 24 y 64 años de edad a los cuales se les calculó HOMA para demostrar la aplicabilidad de este método en el análisis de la insulinoresistencia y función de la célula b en dos etnias diferentes. En esta investigación se comprobó que la IR cuantificada a través del HOMA estuvo fuertemente correlacionada con la concentración de insulina plasmática, así como también se encontraron diferencias significativas entre la IR determinada por HOMA entre mexicanos y blancos no hispánicos, bien sea sanos, con intolerancia a la glucosa y con diabetes. Respecto al HOMA
b cell, se observó una correlación moderada con el dI30/dG30 , así como una relación directamente inversa con el tiempo de evolución de los pacientes que eran diabéticos, es decir, a mayor tiempo de evolución y menor control glucémico, menor valor en el HOMAb cell. En este estudio se concluye que el HOMA provee un modelo muy útil para la cuantificación de la insulinoresistencia y la función de la célula beta en estudios epidemiológicos donde solo se tiene disponible una muestra de suero en ayuno.El Estudio Familiar de Diabetes de Oxford y Northhamptom
Este fue un estudio practicado en padres (564 padres y 192 controles) de pacientes diabéticos tipo 2 (caucásicos) en estos dos condados de Inglaterra(9). Su objetivo fue caracterizar el fenotipo antropométrico, clínico, genético bioquímico y metabólico de los individuos estudiados con el fin de investigar su heterogeneidad y los modelos familiares de diabetes tipo 2.
Los estudios de biología molecular tuvieron como objetivo determinar los Loci genéticos contribuyentes al desarrollo de la diabetes y buscar los genes candidatos. La estimación de la función de la célula b y de insulinosensibilidad se practicó a través del HOMA.
En este estudio se analizaron todas las asociaciones posibles algunas variables que forman parte del síndrome metabólico y HOMA-IR y HOMA
b-cell. Los resultados de este estudio arrojaron una correlación significativa entre el HOMA-IR con el colesterol total, triacilglicéridos y colesterol en las VLDL independientemente del índice de masa corporal de los individuos. El aumento de la presión arterial diastólica y sistólica se correlacionó fuertemente con el HOMA-IR siempre y cuando no se tomara en cuenta el índice de masa corporal.El estudio de la aplicabilidad clínica del HOMA en pacientes diabéticos tipo 2 en Maracaibo, Estado Zulia
En este trabajo se estudiaron 25 individuos diabéticos tipo 2 de ambos sexos que acudían a la consulta de nuestro Centro de investigaciones localizado en la Ciudad de Maracaibo, Venezuela(42). Los criterios diagnósticos de diabetes fueron los definidos por la American Diabetes Association en 1997 y el tiempo promedio de evolución de la enfermedad fue de 7,5 años además, de un control irregular de la enfermedad puesto en evidencia por la historia clínica y niveles de HbA1c elevados. Este grupo de pacientes fue comparado con un grupo control representado por individuos sanos y con un promedio de edad similar al grupo experimental. Los resultados de este trabajo demostraron que los pacientes diabéticos presentaron un HOMA-IR mucho mayor que los pacientes sanos (aunque los sanos también eran algo insulinoresistentes, muy probablemente por el efecto del envejecimiento y el sobrepeso) y un HOMA beta cell menor que los individuos sanos, fenómeno que representa un funcionalismo pancreático alterado. Este estudio, a pesar de haber sido realizado en un grupo pequeño de pacientes ha demostrado la utilidad del HOMA a nivel de la consulta diabetológica diaria.
Estudio preliminar de la aplicabilidad del HOMA en individuos jóvenes sanos con y sin familiares en primer grado con diabetes mellitus tipo 2 en Maracaibo, Estado Zulia
Este es un estudio que se desarrolla en la actualidad en Maracaibo, el cual intenta comprobar alteraciones muy tempranas en la sensibilidad insulinínica y funcionalismo de la célula beta pancreática al contrastar las diferencias a través de HOMA en individuos jóvenes sanos (no obesos) con antecedentes de diabetes (familiares en primer grado) y en individuos jóvenes sanos (no obesos) sin antecedentes familiares de diabetes. Se esperan sus resultados para finales del año 2.000.
Dedicatoria
Este artículo está dedicado a la memoria del Dr. Robert Turner (1938 – 1999) visionario e innovador investigador en el área de la diabetología, en especial, en el desarrollo del United Kingdom Pospective Dabetes Study (UKPDS) y uno de los creadores de los modelos matemáticos HOMA, CIGMA y el modelo de simulación de riesgo de DM tipo 2 basado en los datos del UKPDS.
REFERENCIAS BIBLIOGRÁFICAS
1. Rudenski AS, Matthews DR, Levy JC. Understanding «insulin resistance»: both glucose resistance and insulin resistance are required to model human diabetes. Metabolism 1991; 40(9): 908-917. [ Links ]
2. Turner RC, Matthews DR, Clark A. Pathogenesis of NIDDM-a disease of deficient insulin secretion. Baillieres Clin Endocrinol Metab 1988; 2(2): 327-342. [ Links ]
3. O’Rahilly S, Hattersley A, Vaag A. Insulin resistance as the major cause of impaired glucose tolerance: a self-fulfilling prophesy?. Lancet 1994; 344(8922): 585-589. [ Links ]
4. Edelman SV: Impaired glucose tolerance: A precursor of NIDDM or a disease entity in itself? Diabetes. 1995; 16: 1-5. [ Links ]
5. Bonora E, Targher G, Alberiche M, Bonadonna RC, Saggiani F, Zenere MB, Monauni T, Muggeo M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000; 23(1): 57-63. [ Links ]
6. Hosker JP, Matthews DR, Rudenski AS. Continuous infusion of glucose with model assessment: measurement of insulin resistance and beta-cell function in man. Diabetologia 1985; 28(7): 401-411. [ Links ]
7. Turner RC, Rudenski AS, Hosker JP. CIGMA as a tool in the study of NIDDM. Prog Clin Biol Res 1988; 265: 13-26. [ Links ]
8. Ferrannini E, Mari A. How to measure insulin sensitivity. J Hypertens 1998; 16(7): 895-906. [ Links ]
9. Matthews DR, Hosker JP, Rudenski AS. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28(7): 412-419. [ Links ]
10. Emoto M, Nishizawa Y, Maekawa K, Hiura Y, Kanda H, Kawagishi T, Shoji T, Okuno Y, Morii H. Homeostasis model assessment as a clinical index of insulin resistance in type 2 diabetic Patients treated with sulfunylureas. Diabetes Care 1999; 22(5): 818-822. [ Links ]
11. Petrus DJ, Jackson MW, Kemnitz JW, Finegood DT. Panciera D. Assessing insulin sensitivity in the cat: evaluation of the hyperinsulinemic euglycemic clamp and the minimal model analysis. Res Vet Sci 1998; 65(2): 179-181. [ Links ]
12. Turner RC, Rudenski AS, Matthews DR, Levy JC, O’Rahilly SP, Hosker JP. Application of structural model of glucose-insulin relations to assess beta-cell function and insulin sensitivity. Horm Metab Res Suppl 1990; 24: 66-71. [ Links ]
13. Fukushima M, Taniguchi A, Sakai M, Doi K, Nagasaka S, Tanaka H, Tokuyama K, Nakai Y. Homeostasis model assessment as a clinical index of insulin resistance. Comparison with the minimal model analysis. Diabetes Care 1999; 22(11): 1911-1912. [ Links ]
14. Bavdekar A, Yajnik CS, Fall CH. Insulin resistance syndrome in 8-year-old Indian children: small at birth, big at 8 years, or both? Diabetes 1999; 48(12): 422-429. [ Links ]
15. Himsworth HP, Kerr RB.: Insulin-sensitive and insulin-insensitive types of diabetes mellitus. Clin Sci 1942; 4: 120-152. [ Links ]
16. Ruderman N, Chisholm D, Pi – Sunyer X, et al: The metabolically obese, normal – weight individual revisited. Diabetes.1998; 47: 699-713. [ Links ]
17. Bergman RN, Ader M: Free Fatty Acids and Pathogenesis of Type 2 Diabetes Mellitus. Trends Endocrinol Metab. 2000; 11(9): 351-356. [ Links ]
18. Rudenski AS, Hosker JP, Burnett MA, Matthems DR, Turner RC. The beta cell glucose stimulus-response curve in normal humans assessed by insulin and C-peptide secretion rates. Metabolism. 1988; 37(6): 526-534. [ Links ]
19. Hellman B, Gylfe E, Bergsten P, Grapengiesser E, Lund PE, Berts A, Tengholm A, Pipeleers DG, Ling Z. Glucose induces oscillatory Ca2+ signaling and insulin release in human pancreatic beta cells. Diabetologia. 1994; 37 (Suppl 2): S11-S20. [ Links ]
20. Reis AF, Ye WZ, Dubois-Laforgue D, Bellanne-Chantelot C, Timsit J, Velho G: Association of a variant in exon 31 of the sulfonylurea receptor 1 (SUR1) gene with type 2 diabetes mellitus in French Caucasians. Hum Genet. 2000; 107(2): 138-144. [ Links ]
21. Tsuura Y, Ishida H, Okamoto Y, Tsuji K, Kurose T, Horie M, Imura H, Okada Y, Seino Y. Impaired Glucose Sensitivity of ATP-Sensitive K+ Channels in Pancreatic b-Cells in Streptozotocin-Induced NIDDM Rats. Diabetes. 1992; 41: 861-865. [ Links ]
22. Dunne M, Cosgrove K, Shepherd R, Ammala C. Potassium Channels, Sulphonylurea Receptors and Control of Insulin Release. TEM 1999; 10(4): 146-152. [ Links ]
23. Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998; 21: 197-203. [ Links ]
24. Celi FS, Negri C, Tanner K, Raben N, De Pablo F, Rovira A, Pallardo LF, Martin-Vaquero P, Stern MP, Mitchell BD, Shuldiner AR: Molecular scanning for mutations in the insulin receptor substrate-1 (IRS-1) gene in Mexican Americans with Type 2 diabetes mellitus. Diabetes Metab Res Rev. 2000; 16(5): 370-377. [ Links ]
25. Ottensmeyer FP, Beniac DR, Luo RZ, Yip CC: Mechanism of Transmembrane Signaling: Insulin Binding and the Insulin Receptor. Biochemistry. 2000 Oct10; 39(40): 12103-12112. [ Links ]
26. Van Haeften TW. Glucose tolerance, insulin sensitivity, and the homeostasis model assessment method. Diabetes Care. 1998; 21(4): 673-674. [ Links ]
27. Tai ES, Lim SC, Chew SK, Tan BY, Tan CE. Homeostasis model assessment in a population with mixed ethnicity: the 1992 singapore national health survey. Diabetes Res Clin Pract. 2000; 49(2-3): 159-168. [ Links ]
28. Opara EC, Atwater I, Go VL. Characterization and control of pulsatile secretion of insulin and glucagons. Pancreas. 1988; 3(4): 484-487. [ Links ]
29. Polonsky KS, Sturis J, Van Cauter E. Temporal profile and clinical significance of pulsatil insulin secretion. Horm Res. 1998; 49(3-4): 178-184. [ Links ]
30. Jones CN, Pei D, Staris P.Alterations in the glucose-stimulated insulin secretory dose-response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab. 1997; 82(6): 1834-1838. [ Links ]
31. Rudenski AS, Matthews DR, Levy JC. Understanding «insulin resistance»: both glucose resistance and insulin resistance are required to model human diabetes. Metabolism. 1991; 40(9): 908-917. [ Links ]
32. Brun JF, Raynaud E, Mercier J. Homeostasis model assessment and related simplified evaluations of insulin sensitivity from fasting insulin and glucose. Diabetes Care. 2000; 23(7): 1037-1038. [ Links ]
33. Rudenski AS, Hadden DR, Atkinson AB, Kennedy L, Matthews DR, Merrett JD, Pockaj B,Turner RC. Natural history of pancreatic islet B-cell function in type 2 diabetes mellitus studied over six years by homeostasis model assessment. Diabet Med. 1988; 5(1). [ Links ]
34. Levy J.C. Évaluation de l‘insulinosensibilité: Les modèles homa et cigma. Flamarion medecine-Scienses. Actualitès nephrologiques. 1998. [ Links ]
35. Fukushima M, Taniguchi A, Sakai M. Homeostasis model assessment as a clinical index of insulin resistance. Comparison with the minimal model analysis. Diabetes Care. 1999; 22(11): 1911-1912. [ Links ]
36. Levy JC; Matthews DR; Hermans MP. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998; 21(12): 2191-2192. [ Links ]
37. Pacino G, Finegood D, Bergman N. A minimal model-Based glucose clamp yielding insulin sensitivity independent of glycemia. Diabetes. 1982: 31. [ Links ]
38. Doberne L, Greenfield M, Schulz B, Reaven G. Enhanced glucose utilization during prolonged glucose clamp studies. Diabetes. 1981; 30. [ Links ]
39. Bergman R. Toward Phisiology understanding of glucose tolerance: Minimal model approach. Diabetes. 1989; 38. [ Links ]
40. Haffner SM; Kennedy E; Gonzalez C; Stern MP; Miettinen H. A prospective analysis of the HOMA model. Diabetes Care. 1996; 19(10): 1138-1141. [ Links ]
41. Haffner SM; Miettinen H; Stern MP. The homeostasis model in the San Antonio Heart Study. MP Diabetes Care. 1997; 20(7): 1087-92. [ Links ]
42. Bermúdez V, Arias N, Souki A, Ambard M, Medina M, Núñez M, Andrade J, Lemus M, Leal Elliuz, Arria M, Bermúdez F, Cano C y Contreras F. Aplicación de un modelo de Valoración Homeostática en pacientes diabéticos tipo 2. Arch Farm y Terap. 2000; 19(1): 53-57. [ Links ]