








Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkGaceta Médica de Caracas
Print version ISSN 0367-4762
Gac Méd Caracas vol.110 no.1 Caracas Jan. 2002
Enfermedades priónicas en humanos
Drs. Alipio A. Hernández F., Ghislaine Céspedes C., Jesús E. González A.
Sección de Neuropatología. Instituto Anatomopatológico "Dr. José A. ODaly". Universidad Central de Venezuela. Caracas, Venezuela.
RESUMEN
Las enfermedades priónicas comprenden entidades neurodegenerativas fatales en humanos y animales. La extensión de la encefalopatía espongiforme bovina en diferentes países de Europa y la aparición de la nueva variante de la enfermedad de Creutzfeldt-Jakob en humanos por la ingesta de productos bovinos contaminados, hacen de las enfermedades priónicas un verdadero problema de salud pública mundial en la actualidad. En este artículo se tratan los aspectos generales de estas enfermedades y se presenta la casuística venezolana acumulada durante los últimos 30 años.
Las enfermedades priónicas (EP) comprenden entidades neurodegenerativas en humanos y animales (Cuadro 1) que son producidas por el metabolismo aberrante de la proteína priónica (PrP) y se caracterizan por un período de incubación prolongado, transmisibilidad a animales de experimentación y evolución clínica fatal (1-3). La forma esporádica o idiopática de la enfermedad de Creutzfeldt-Jakob (ECJ) comprende el 85-90% de las EP en humanos, con una incidencia mundial de 1-2 nuevos casos por millón de habitantes por año y una distribución geográfica amplia (2-4). Suele presentarse alrededor de los 60 años de edad bajo la forma clásica de un cuadro demencial rápidamente progresivo asociado a mioclonos y electroencefalograma (EEG) periódico (5-7). Histopatológicamente, existe afectación difusa de la sustancia gris superficial y profunda del sistema nervioso central (SNC), con degeneración espongiforme, astrogliosis y pérdida neuronal en ausencia de infiltrado inflamatorio (2,3,6). La sobrevida suele ser de un año y no existe hasta la actualidad un tratamiento médico efectivo contra la enfermedad (2,5,6).
Enfermedades priónicas
(Prusiner SB. Brain Pathol 1998;8:499-513)
| En humanos | En animales |
| Infecciosas* | Scrapie de ovejas y cabras |
| Kuru | Encefalopatía espongiforme bovina |
| ECJ latrogénica | Encefalopatía transmisible del visón |
| Nueva Variante | Enfermedad de desgaste crónico de venados y alces |
| Hereditarias | Encefalopatía espongi forme felina |
| ECJ Familiar | Encefalopatía de ungulados exóticos (gran kudu, nyala, oryx) |
| Enfermedad de Gerstmann Sträussler-Scheinker | |
| Insomnio familiar fatal | |
| Idiopática | |
| ECJ Esporádica |
El Kurú es una enfermedad prevalente de la tribu Fore de Nueva Guinea y prácticamente ha desaparecido con el cese de sus rituales canibalistas (ingestión de cerebro y ojos humanos). La ECJ iatrogénica se produce por transmisión de priones a través de transplantes de córnea, neurocirugía y electroencefalografía estereotáxica con el uso de instrumentos y objetos contaminados, injertos de duramadre e inoculación de hormona de crecimiento y gonadotrofina coriónica humana cadavérica obtenida de humanos con la enfermedad. La nueva variante de la ECJ es ocasionada por el consumo de productos bovinos contaminados con la EEB.
En el pasado, las EP fueron conocidas como enfermedades por virus lento, dada la sospecha de que respondían a un agente viral con un período de incubación muy prolongado (8-10). Ante la imposibilidad de demostrar tal agente, se adoptó el término de encefalopatía espongiforme transmisible (EET) por el criterio histopatológico diagnóstico de estas enfermedades (degeneración espongiforme) y la transmisibilidad a animales de experimentación mediante el uso de inóculos de tejido cerebral infectado (8,11,12). Desde hace unas dos décadas, ya descubierta y caracterizada molecularmente la PrP como agente causal de las EET, se emplea el término general de EP (8,11,13).
PrP Celular y Prión.
La isoforma normal de la PrP (PrP celular o PrPC) es expresada como glicoproteína de membrana por las neuronas de todos los mamíferos y algunas aves (2,12,14). En humanos, está constituida por 253 aminoácidos y es codificada por el gen de la (PRNP) localizado en el cromosoma 20 (2,10,15). También ha sido aislada en células de tejidos extraneurales (14,16,17). Su función biológica es desconocida (9,10,12,14), aunque se ha señalado puede tratarse de una molécula de adhesión, señalización celular o receptor de membrana (2,12,14,18). Estudios recientes indican que la PrPC actúa como una proteína unidora de cobre y de esta manera ejerce un rol protector de la sinapsis al prevenir la toxicidad por cobre y contrarrestar el estrés oxidativo (2,19,20).
El término "prión" denota la partícula proteinácea infecciosa carente de ácido nucléico que produce las EP en humanos y animales (3,12,21). Esta PrP infecciosa, denominada PrP del scrapie en animales (PrPSe) y de la ECJ en humanos (PrPECJ), difiere estructuralmente de cualquier microorganismo conocido, incluyendo a virus y viroides (PrPECJ), difiere estructuralmente de cualquier microorganismo conocido, incluyendo a virus y viroides, siendo resistente al calor (hasta 100ºC o 212ºF), congelamiento, radiación ionizante y ultravioleta, desinfección química convencional y fijación prolongada en formol (2,14,22). También se ha señalado que la PrPECJ puede persistir durante años en el ambiente (23).
La PrPC y la PrPECJ pueden ser similares desde el punto de vista bioquímico (9,10,12). Son codificadas por el mismo gen, sufren glicosilación en sitios similares y presentan un residuo fosfatidilinositol idéntico para el anclaje a la membrana celular (9,10,12,24). La diferencia fundamental entre ambas proteínas radica en su estructura terciaria (3,9,24).

Figura 1. Modelo de replicación priónica: mediante acoplamiento o dimerización, la PrPECJ induce la conversión postranslacional de la PrPC en otra molécula de PrP.
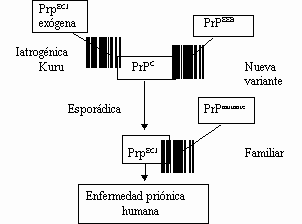
Figura 2. Vías de conversión de la PrPC en PrP ECJ en las diferentes variantes de la enfermedad priónica humana. (Ironside JW. J Pathol 1998;186:227-234.
La PrPC es más hidrofóbica por tener un contenido rico en hélices (42%) y bajo en láminas ß (3%), en contraste con la PrPECJ que tiene un mayor contenido de láminas ß (43%) y menor de hélices (30%) 3,12,25 (Figura 1). Esta disposición espacial diferente de la PrPECJ le confiere resistencia parcial a la digestión con proteasas, lo que explicaría su depósito anormal en los lisosomas secundarios de las neuronas afectadas (1,3,12).
Replicación priónica y barreras de especies
Toda vez que una molécula de PrPECJ se pone en contacto con una molécula de PrPC provoca pérdida de hélices y ganancia de láminas ß en esta última, convirtiéndola en un prión infectante (1,3,12) (Figura 1). A este fenómeno biológico de autoreplicación proteica se le conoce como transformación postranslacional, siendo la PrPC y la PrPECJ isiformas conformacionales (2,3,12). Sea cual sea la forma mediante la cual se adquieren los priones infectantes, el mecanismo de autoreplicación de la PrPECJ es idéntico (8,26). En humanos, la PrPECJ puede originarse por mutaciones en el PRNP (priones endógenos), por conversión espontánea de la PrPC en PrPECJ o por una transformación postranslacional de la PrPC ocasionada por priones exógenos (8,26) (Figura 2). Una vez formada en la membrana plasmática de la célula, la PrPECJ sigue la ruta metabólica de la PrPC: es introducida al citoplasma de las neuronas a través de endocitosis mediada por receptor, continúa replicándose en todo el sistema endocíticolisosomal y no es degradada en los lisosomas secundarios, donde finalmente se acumula por tener una vida media infinita, aunque una parte de la PrPECJ también puede pasar al aparato de Golgi y vía vesículas transportadoras ser devuelta a la membrana plasmática (12,24,27). Es importante señalar que la localización subcelular precisa donde tiene lugar la acumulación de la PrPECJ sigue siendo motivo de investigación (12,28). Existe evidencia científica acerca de la existencia de un transporte de PrPC y PrPECJ a todo lo largo del axón de las neuronas y, en sistemas de cultivo celular, se ha demostrado que la PrPECJ se acumula fundamentalmente en los lisosomas secundarios de las células o en estructuras relacionadas (cuerpos multivesiculares y estructuras túbulo-reticulares) (12,27). Dado el patrón granular fino de tipo sináptico que se observa con el marcaje de la PrPECJ mediante inmonohistoquímica e inmunomicroscopia electrónica (25,28), algunos autores han señalado que la diseminación de los priones en el SNC no ocurre por difusión simple una vez producida la muerte y desintegración celular, sino que los priones se propagan por continuidad entre las células infectadas siguiendo sus sinapsis y afectando de esta manera vías neuroanatómicas bien específicas (14,28).
Los ratones Prnp 0/0 (que no expresan PrPC) no desarrollan scrapie ni propagan los priones luego de su inoculación con PrPSe (11,12,29). La ausencia de la PrPC no representa la causa de la disfusión neuronal en la EP, aunque es necesaria como substrato para que ocurra la replicación del prión (2,14). Por otra parte, la transmisión de la EP es más efectiva en la medida en que estructuralmente son más parecidas la PrP infecciosa y la PrPC del huésped (12,24,30). En condiciones normales, el prión que produce el scrapie en hamsters sirios no provoca la enfermedad en ratones CD-1 (12,31). Sin embargo, la creación de ratones trangénicos hídricos que expresan la PrPC del hamster sirio y no la del ratón (ratones ShaPrP/Prnp0/0), desarrollan scrapie luego de la inoculación con PrPSC de hamster, pero no con la PrPSC de ratón (3,12). De esto se concluye que los priones que provocan enfermedad en una determinada especie animal no son patógenos para otras especies, debido a diferencias en la secuencia de aminoácidos entre la PrPC y la PrPSe (3,24). A esto se refiere la "barrera de especies"(12,24,31) y explica por que los humanos no han adquirido la EP a partir del scrapie de ovejas y cabras en regiones del mundo donde esta EET animal es frecuente (4,9). La única EP animal a la cual el hombre es susceptible es la encefalopatía espongiforme bovina (EEB), lo que pudiera explicarse por el hecho de que la PrPC bovina contiene regiones estructuralmente muy parecidas a dominios claves de la PrPC humana (1,3,32).
En animales de experimentación, el "fenotipo" de la EP está determinado por el período de incubación de la enfermedad y el patrón de lesión cerebral que se produce (12,33,34). Según la teoría del "prión único" o "modelo priónico de un componente", se esperaría que el fenotipo de la EP fuera el mismo para una determinada especie una vez ocurrida la infección priónica, puesto que la autoreplicación proteica tendría lugar a partir de la PrPC del huésped (3,9,35). Sin embargo, el fenotipo de la EP (animal o humana) es variable y depende de la influencia de 3 factores: el tipo de prión, la vía de la infección y el genotipo del huésped (12,35,36).
EP y tipos de priones
En ratones transgénicos híbridos que expresan al mismo tiempo PrPC de hamster sirio y de ratón, la inoculación con priones de hamster sirio (Sc237) conduce a la producción de PrPSc de hamster sirio, en tanto que los priones de ratón (RML) inducen la formación de PrPSc de ratón (11,12,21). Este resultado es el esperado, si se toma en cuenta que la homología estructural PrPC/PrPSc es necesaria para una replicación efectiva del prión (12,21,31). Lo interesante del experimento es que los dos tipos de priones dan lugar a diferentes patrones de lesión cerebral: la PrPSc de hamster cursa con formación de placas amiloides y no afecta a la sustancia blanca cerebral, en tanto que la PrPSc de ratón afecta a la sustancia blanca y no se asocia a placas amiloides (11,12,37). De todo esto se desprende que existen diferentes "tipos o clases" de priones que determinan diferentes fenotipos de EP (33,36,38,39). Se ha demostrado además que un determinado fenotipo de EP se mantiene estable a través de la inoculación sucesiva de priones en diferentes huéspedes, aún ante la presencia de PrPC con una secuencia de aminoácidos diferente (1,12,21,34). De esta manera, se puede señalar que la PrPC juega un rol fundamental en la replicación priónica, pero es en definitiva el prión infectante el que determina el tipo de enfermedad que se desarrollará (3,12,36,40). Esto fue lo que hizo pensar durante mucho tiempo que las EP eran de etiología viral, puesto que el prión actúa como un virus llevando consigo la información necesaria para provocar un fenotipo determinado de EP (3,24,33). Ante la ausencia de un ácido nucléico, se cree que esta información reside en el estado conformacional y grado de glicosilación del prión (39-41).
EP y vía de infección
El período de incubación y las manifestaciones clínicas de la EP también son influenciados por la vía mediante la cual se produce la infección priónica (38,42). Esto ha sido demostrado en pacientes con la ECJ iatrogénica, en quienes es posible determinar el período de incubación de la enfermedad. En aquellos casos en los cuales la infección priónica se produce por vía central o intracraneal (neurocirugía, EEG estereotáxica, transplantes de córnea e injertos de duramadre), la demencia representa la manifestación clínica inicial y predominante de la enfermedad, con un período de incubación variable entre los 1,5 y 7 años (43). En contraste, aquellos pacientes que se infectan por vía periférica (uso intramuscular de hormona de crecimiento o gonadotrofina coriónica humana cadavérica contaminada con priones) inician el cuadro con trastornos cerebelosos y muestran un período de incubación mucho más prolongado (alrededor de 12 años) (43).
La vía de la infección también determina la efectividad con la cual los priones provocan la EP (9). En comparación con la vía central (intracraneal), se requiere de un mayor número de unidades infecciosas para producir una transmisión efectiva en animales de experimentación luego de la inoculación de tejidos infectados por rutas periféricas (intraperitoneal, intramuscular, oral, etc.) (9). Así por ejemplo, se ha establecido que la infección intragástrica es aproximadamente 1/40 000 veces tan efectiva como la infección intracerebral directa para provocar EP (9) y que la simple ingestión de priones (ruta oral) es 109 veces menos eficiente en producir la enfermedad que la vía central de la infección (44).
EP y Genotipo del huésped
Existen 20 mutaciones del PRNP que producen EP familiar siguiendo un patrón de herencia autosómico dominante, de las cuales 13 ocurren por sustitución puntual de un aminoácido por otro y 7 por inserción de pares de bases en la región del octapéptido repetido del PRNP (2,3,15). La EP familiar más frecuente (90% de los casos) es la ECJ familiar que se produce por una sustitución de ácido glutámico por lisina en el codon 200 del PRNP, la cual es designada bajo la nomenclatura E200K, 200Lis o ECJ 200 (45). El fenotipo de la EP familiar varía según el tipo de mutación que ocasiona la enfermedad (2,3,15,37). Las mutaciones puntuales D178N (con valina en el codon 129 del alelo mutado), V108I, T183A, E200K, R208K, V210I y M232R dan lugar a la variante familiar de la ECJ (2,8,46). La enfermedad de Gertsmann-Schinker (GSS) es producida por las mutaciones A117V, P102L, P105L, Y145*, F198S y Q217R (2,3,21). Las mutaciones por inserción de octapéptidos repetidos en la PrPC están relacionadas con manifestaciones clínicas atípicas, una duración prolongada de la enfermedad y grados variables de cambio espongiforme (8,45,47).
Una de las particularidades más fascinantes en el estudio de la EP familiar tiene que ver con los "polimorfismos" en el PRNP, los cuales determinan no soló el fenotipo de la EP familiar sino también el grado de susceptibilidad que se tiene ante la infección priónica (3,48-50). El ejemplo clásico está representado por la relación existente entre uno de los tipos de ECJ familiar y el insomnio familiar fatal (IFF) (2,3). La mutación Puntual D178N representa la lesión molecular en ambas entidades, pero un polimorfismo en el codon 129 del PRNP determina cuál de ellas se desarrollará (2,3,12). Si en el alelo mutado del PRNP el codon 129 es ocupado por metionima (M) se produce IFF (ausencia de demencia y compromiso casi exclusivo del tálamo), en tanto que si es valina (V) la que ocupa tal posición se desarrolla ECJ familiar (demencia progresiva y afectación difusa de la corteza cerebral y núcleos subcorticales) (2,3,12). Este polimorfismo también se ha estudiado en la población normal y en pacientes con otras variantes de la ECJ. El 50% de las personas normales muestran un estado heterocigoto M/V en el codon 129 del PRNP, seguido por los homocigotos M/M (37%) y V/V (13%) (11,12,51). Si se toma en cuenta que el 80% de los pacientes con la ECJ esporádica (12,25,43) y todos aquellos con la nueva variante de la ECJ (nvECJ) (51-53) son homocigotos M/M en el codon 129 del PRNP, se puede concluir entonces que la condición de heterocigoto proporciona cierta "protección" contra la infección priónica, en tanto que la condición de homocigoto M/M otorga un mayor grado de susceptibilidad para contraer la enfermedad (2,49,54).
PrP y tejidos extraneurales
En humanos, la PrPC ha sido aislada en tejidos extraneurales como pulmones, hígado, riñones, aparato reproductor, glándulas salivales, sistema linfoide, corazón, músculo esquelético, páncreas y diferentes componentes de la sangre (1,55,56). Si se toma en cuenta que la PrPC representa el substrato sobre el cual se multiplican los priones, se supone entonces que todos los tejidos extraneurales de un paciente con EP pueden ser potencialmente infecciosos. De hecho, la EP humana se ha transmitido a primates con el uso de inóculos de tejido pulmonar, hepático, renal, esplénico, de ganglios linfáticos, sangre y orina (42,57,58). Además, en pacientes con la nvECJ los priones infecciosos se han aislado desde las amígdalas palatinas y todo el tejido linfático (2,16).
Dentro de la patogenia de la EP, el rol que juegan las células dendríticas foliculares del sistema linfático en casos de infección por vía periférica ha sido bien establecido (17,55,59,60). En ratones inoculados con la PrPSc por vía intraperitoneal, intravenosa y subcutánea se ha demostrado un paso previo de replicación priónica en el tejido linfático (especialmente en el bazo, placas de Peyer y amígadalas palatinas) antes de producirse la afectación del SNC (16,17,59). Este paso previo de replicación parece facilitar la entrada de los priones al SNC (14,59,60). De hecho, se ha encontrado que los ratones esplenectomizados y posteriormente inoculados con priones (vía periférica) muestran un período de incubación a la enfermedad mucho más prolongado que aquellos ratones no esplenectomizados (55). Lo que todavía sigue siendo un enigma es el mecanismo mediante el cual los priones infecciosos que se multiplican en el bazo invaden posteriormente al SNC, aunque se piensa lo hacen vía fibras nerviosas autonómicas y médula espinal (14,16,61).
EEB y nueva variante de la ECJ
La nvECJ es ocasionada por el consumo de tejidos infectados con la PrP de la EEB y está relacionada con la epidemia de las "vacas locas" ocurrida en Gran Bretaña entre 1989-92 (32,38,62,63). A diferencia de la ECJ clásica, afecta a pacientes jóvenes (alrededor de los 30 años) y su evolución clínica es de mayor duración (5,51,53,64). Se manifiesta inicialmente por síntomas psiquiátricos (cambios de conducta, ansiedad o depresión) y trastornos sensoriales periféricos (51,53,64,65). La demencia, ataxia progresiva y movimientos anormales (mioclonos o corea) se desarrollan tardíamente y no se observa el EEG periódico característico (53,64-66). Aparte de los hallazgos habituales, el examen neuropatológico muestra abundantes placas tipo Kuru y "floridas" en la corteza cerebral y cerebelosa (2,34,53). No existen mutaciones en el PRNP y todos han sido homocigotos M/M en el codon 129 (53,64,67,68). Hasta el 2000, 91 casos de la nvECJ habían sido confirmados (87 en el Reino Unido, 3 en Francia y 1 en Irlanda) (51,69) y, aunque han pasado más de 10 años desde la epidemia de las "vacas locas", no se conoce a ciencia cierta el verdadero impacto poblacional que tendrá en el futuro el consumo del ganado infectado durante los años de la misma (1,51,70,71), con el agravante de los nuevos brotes de EEB que han aparecido recientemente en diferentes países de Europa (13,72,73).
Adelantos en el tratamiento de la EP
En la actualidad, múltiples modalidades de tratamiento anti-EP están siendo objeto de evaluación experimental (26,74). Como descubrimiento prometedor, los péptidos bloqueadores de láminas ß han demostrado en cultivos celulares y en animales de experimentación su capacidad para inhibir la replicación de la PrPSc y revertir los cambios estructurales ocurridos durante la transformación postranslacional de la PrPSc (75-77). Otros compuestos con un efecto similar comprenden colorantes aniónicos (rojo Congo) (78), polianiones sulfatados (26,79), antibióticos polienos (anfotericina B y derivados) (80-82), antraciclinas (83), tetrapirroles cíclicos (84,85), poliaminas ramificadas (86), quinacrina e inhibidores de la cisteína proteasa (87). Sin embargo, la eficacia de la mayoría de estos compuestos ha sido mayor sólo cuando son administrados en el momento en que ocurre la infección priónica (lo cual es difícil de determinar en humanos) y los resultados obtenidos han variado según el tipo de prión y otros factores inherentes al compuesto (dosis, ruta y tiempo de administración, etc.) (84,86,87). La citotoxicidad, metabolismo fisiológico de la droga, generación de respuesta inmune y permeabilidad de la barrera hematoencefálica, son sólo algunos de los obstáculos que in vivo deben salvar las investigaciones futuras antes del inicio de la experimentación de estas modalidades terapéuticas en seres humanos (76,85).
ECJ en Venezuela
Un total de 16 casos de la ECJ esporádica se han confirmado en Venezuela durante el período 1972-2000 (Cuadro 2 y Figura 3). Con la excepción de un paciente cuyo diagnóstico estuvo basado en datos clínicos y electroencefalográficos, la ECJ esporádica fue confirmada por examen neuropatológico en el resto de los pacientes (biopsia cerebral en 3 casos y autopsia en 12) e inmunohistoquímica en 5 casos. La mayoría de los pacientes (12 casos) fueron estudiados en el Instituto Anatomopatológico de la Universidad Central de Venezuela y los restantes en la Universidad del Zulia. Los primeros 14 pacientes del Cuadro 2 han sido objeto de publicaciones previas (88-91).
ECJ Esporádica: casuística venezolana
| Caso | Sexo/Edad | Año de muerte |
| 1 | F/50 | 1972 |
| 2 | M/42 | 1979 |
| 3 | M/60 | 1982 |
| 4 | F/61 | 1986 |
| 5 | M/75 | 1986 |
| 6 | F/64 | 1989 |
| 7 | F/42 | 1990 |
| 8 | F/64 | 1991 |
| 9 | M/32 | 1991 |
| 10 | F/48 | 1994 |
| 11 | M/60 | 1994 |
| 12 | F/72 | 1995 |
| 13 | M/69 | 1998 |
| 14 | F/63 | 1999 |
| 15 | F/48 | 2000 |
| 16 | F/71 | 2000 |
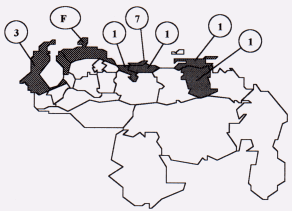
Figura 3. Distribución Geográfica de los Casos de ECJ en Venezuela (período 1972-2000)*
*El número de casos por estado se indica dentro de los círculos. La F indica el grupo familiar con la ECJ T183A del Estado Falcón. Dos de los pacientes con ECJ esporádica no aparecen en el mapa: uno procedía de Chile y falleció poco después de su llegada al país; en el otro paciente no se informó su lugar de residencia.
El promedio de edad de los pacientes con ECJ esporádica fue de 57,6 años (rango de 35-72 años) y el intervalo de tiempo hasta la muerte de 5,6 meses (rango de 1-24 meses). Todos desarrollaron un síndrome demencial rápidamente progresivo asociado a trastornos piramidales/extrapiramidales, mioclonos y cambios en el EEG. El examen neuropatológico reveló degeneración espongiforme, astrocitosis y pérdida neuronal en la sustancia gris de la corteza cerebral, núcleos basales y cerebelo. La distribución geográfica según el lugar de residencia de los pacientes está representada en la Figura 3. La mayoría de los casos eran residentes de la Región Capital (7 casos) y 3 pacientes procedían del Estado Zulia. Los estados Trujillo, Aragua, Miranda y Sucre están representados por un caso cada uno. Uno de los pacientes era procedente de Chile y en otro no se conoció su lugar de residencia. Como puede notarse en la Figura 3, existe una vasta extensión del territorio nacional donde no se han registrado casos de la ECJ y sobre este respecto se sospecha un subregistro por desconocimiento de la enfermedad y/o falta de confirmación neuropatológica de casos sospechosos. En el Cuadro 2 se observa que los casos de ECJ esporádica se han ido presentando paulatinamente durante los últimos 30 años y, si se considera que la población general del país ha variado entre los 15 y 22 millones de habitantes durante este período de tiempo, se puede concluir entonces que la frecuencia de la ECJ esporádica en Venezuela es baja al compararla con las cifras informadas en otros países (1-2 casos nuevos por millón de habitantes por año).
Aparte de los casos esporádicos antes descritos, en el Estado Falcón se detectaron 2 hermanos con la ECJ familiar, en quienes fue posible confirmar la mutación puntual T183A en el PRNP (92). Como antecedentes de importancia en estos pacientes, la madre y un hermano habían fallecido previamente por la enfermedad y otro hermano (todavía vivo) está siendo objeto de seguimiento clínico.
Con base en las manifestaciones clínicas y hallazgos neuropatológicos de los pacientes estudiados hasta la fecha, se concluye que no existen en Venezuela casos relacionados con la EEB. Además, según información recabada en la Facultad de Ciencias Veterinarias de la Universidad Central de Venezuela, en el Instituto de Investigaciones Agrícolas del Ministerio de Ciencias y Tecnología y en el Servicio Autónomo de Sanidad Agropecuaria del Ministerio de la Producción y el Comercio, no se ha registrado hasta este momento ningún caso de EP animal en nuestro país.
REFERENCIAS
1. Prusiner SB. Prion diseases and the BSE crisis. Science 1997;278:245-250. [ Links ]
2. Ironside JW. Prion diseases in man. J Pathol 1998;186:227-234. [ Links ]
3. Prusiner SB. The prion diseases. Brain Pathol 1998;8:499-513. [ Links ]
4. Brown P, Cathala F, Raubertas RF, Gajdusek DC, Castaigne P. The epidemiology of Creutzfeldt - Jakob disease: Conclusion of a 15 - year investigation in France and review of the world literature. Neurology 1987;37:895-904. [ Links ]
5. World Health Organization. Public health issues and clinical and neurological characteristics of the new variant of Creutzfeldt-Jakob disease and other human and animal transmissible spongiform encephalopathies: memorandum from two WHO meetings. Bull World Health Organ 1996;74:453-463. [ Links ]
6. Kretzchmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol 1996;53:913-920. [ Links ]
7. Will R, Zeidler M. Diagnosing Creutzfeldt-Jakob disease [letter]. BMJ 1996; 313: 833-834. [ Links ]
8. Lantos PL. From slow virus to prion: A review of transmissible spongiform encephalophaties. Histophatology 1992;20:1-11. [ Links ]
9. Haywood AM. Transmissible spongiform encephalophaties. N Engl J Med 1997;337:1821-1828. [ Links ]
10. Brown P, Bradley R. 1755 and all that: A historical primer of transmissible spongiform encephalopathy. BMJ 1998;317:1688-1692. [ Links ]
11. Prusiner SB, Hsiao KK. Human prion diseases. Ann Neurol 1994;35:385-395. [ Links ]
12. DeArmond SJ, Prusiner SB. Etiology and pathogenesis of prion diseases. Am J Pathol 1995;146:785-811. [ Links ]
13. Collec JG, Bradley R. BSE: A decade on-part 1. Lancet 1997;349:636-641. [ Links ]
14. Aguzzi A, Klein MA, Montrasio F, Pekarik V, Brandner S, Furukawa H, et al. Prions: Pathogenesis and reserve genetics. Ann NY Acad Sci 2000;920:140-157. [ Links ]
15. Goldmann W. PrP gene and its association with spongiform encephalophaties. Br Med Bull 1993;49:839-859. [ Links ]
16. Brandner S, Klein MA, Frigg R, Pekarik V, Parizek P, Raeber A, et al. Neuroinvasion of prions: Insights from mouse models. Exp Physiol 2000;85:705-712. [ Links ]
17. McBride PA, Eikelenboon P, Kraal G, Fraser H, Bruce ME. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol 1992;168:413-418. [ Links ]
18. Reilly CE. Nonpathogenic prion protein (PrPc) acts as a cell-surface signal transducter. J Neurol 2000;247:819-820. [ Links ]
19. Brown DR. Prion and prejudice: Normal protein and the synapse. Trends Neurosci 2001;24:85-90. [ Links ]
20. Wong BS, Pan T, Liu T, Li R, Petersen RB, Jones IM, et al. Prion disease: A loss of antioxidant function? Biochem Biophys Res Commun 2000;275:249-252. [ Links ]
21. Prusiner SB. Molecular biology of prion disease. Science 1991;252:1515-1522. [ Links ]
22. Van der Valk P. Prion diseases: What will be next? J Clin Pathol 1998;51:265-269. [ Links ]
23. Bell JE, Ironside JW. Neuropathology of spongiform encephalophaties in humans. Br Med Bull 1993;49:738-777. [ Links ]
24. Prusiner SB. Transgenetics and cell biology of prion diseases: Investigations of PrPSc synthesis and diversity. Br Med Bull 1993;49:873-912. [ Links ]
25. Tateishi J. Prion diseases. Microbiol Immunol 1995;39:923-928. [ Links ]
26. Head MW, Ironside JW. Inhibition of prion-protein conversion: A therapeutic tool? Trends Microbiol 2000;8:6-8. [ Links ]
27. Lazlo L, Lowe J, Self T, Kenward N, Landon M, Mcbride T, et al. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J Pathol 1992;166:333-341. [ Links ]
28. Kitamoto T, Shin R-W, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, et al. Adnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous in patiens with Creutzfeldt-Jakob disease. Am J Pathol 1992;140: 1285-1294. [ Links ]
29. Weissmann C, Büeler H, Fischer M, Sailer A, Aguzzi A, Aguet M. PrP-deficient mice are resistant to scrapie. Ann NY Acad Sci 1994:724:235-240. [ Links ]
30. Collinge J, Palmer MS, Sidle KCL, Hill AF, Gowland I, Meads J, et al. Unaltered susceptibility to BSE in transgenic mice expressing human prion. Nature 1995;378:779-783. [ Links ]
31. DeArmond SJ, Prusiner SB. Prion protein transgenes and the neuropathology in prion diseases. Brain Pathol 1995;5:77-89. [ Links ]
32. Collee JG, Bradley R. BSE: A decade on-part 2. Lancet 1997;349:715-721. [ Links ]
33. Aguzzi A, Weissmann C. A suspicious signature. Nature 1996;383:666-667. [ Links ]
34. Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that "new variant" CJD is caused by BSE agent. Nature 1997;389:498-501. [ Links ]
35. Weissmann C, Büeler H, Sailer A, Fischer M, Aguet M, Aguzzi A. Role of PrP in prion diseases. Br Med Bull, 1993;49:995-1011. [ Links ]
36. Bruce ME. Scrapie strain variation and mutation. Br Med Bull 1993;49:822-838. [ Links ]
37. DeArmond SJ. Overview of the transmissible spongiform encephalophaties: Prion protein disorders. Br Med Bull 1993;49:725-737. [ Links ]
38. Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of "new variant" CJD. Nature 1996;383:685-690. [ Links ]
39. Aucouturier P, Kascsak RJ, Frangione B, Wisniewski T. Biochemical and conformational variability of human prion strains in sporadic Creutzfeldt-Jakob disease. Neurosci Lett 1999;274:33-36. [ Links ]
40. Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in ß-sheet conformations of abnormal prion protein. J Biol Chem 1996;48:32230-32235. [ Links ]
41. Parchi P, Capellari S, Ghen SG, Petersen RB, Gambetti P, Kropp N, et al. Typing prion isoforms [letter]. Nature 1997;386:232-233. [ Links ]
42. Brown P, Gibbs CJ, Rodgers-Jonson P, Asher DM, Sulima MP, Bacote A, et al. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmited disease. Ann Neurol 1994;35:513-529. [ Links ]
43. Will RG. Epidemiology of Creutzfeldt-Jakod disease. Br Med Bull 1993;49:960-970. [ Links ]
44. Diringer H, Beekes M, Oberdieck U. The nature of the scrapie agent: The virus theory. Ann NY Acad Sci 1994;724:246-258. [ Links ]
45. Richardson EP, Masters CL. The nosology of Creutzfeldt-Jakob disease and conditions related to the accumulation of PrPCJD in the nervous system. Brain Pathol 1995;5:33-41. [ Links ]
46. Hsiao KK. The genetics and transgenetics of human prion disease. Ann NY Acad Sci 1994;724:241-245. [ Links ]
47. Skworc KH, Windl O, Schulz-Schaeffer WJ, Giese A, Bergk J, Nägele A, et al. Familial Creutzfeldt-Jakob disease with a novel 120-bp insertion in the prion protein gene. Ann Neurol 1999;46:693-700. [ Links ]
48. Tateishi J, Kitamoto T. Inherited prion diseases and transmission to rodents. Brain Pathol 1995;5:53-59. [ Links ]
49. Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991;352:340-342. [ Links ]
50. Zimmermann K, Turecek Pl, Schwarz HP. Genotyping of the prion protein gene at codon 129. Acta Neuropathol 1999;97:355-358. [ Links ]
51. Brown P, Will RG, Bradley R, Asher DM, Detwiler L. Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: Background, evolution, and current concerns. Emerg Infect Dis URL: http://www.cdc.gov/ ncidoc/eid/vol7no1/brown. htm. [ Links ]
52. Cervenáková L, Goldfarb LG, Garruto R, Lee H-S, Gajdusek DC, Brown P. Phenotype-genotype studies in kuru: Implications for new variant Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 1998;95:13239-13241. [ Links ]
53. Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000;47:575-582. [ Links ]
54. Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet 1991;337:1441-1442. [ Links ]
55. Mabbott NA, Farquhar CF, Brown KL, Bruce ME. Involvement of the immune system in TSE pathogenesis. Immunology Today 1998;19:201-203. [ Links ]
56. Brown P, Cervenáková L, Diringer H. Blood infectivity and the prospects for a diagnostic screenig test in Creutzfeldt-Jakob disease. J Lab Clin Med 2001;137:5-13. [ Links ]
57. Manuelidis EE, Kim JH, Mericangas JR, Manuelidis L. Transmission to animals of Creutzfeldt-Jakob disease from human blood [letter]. Lancet 1985;II:896-897. [ Links ]
58. Tateishi J. Transmission of Creutzfeldt-Jakob disease from human blood and urine into mice [letter]. Lancet 1985;II:1074. [ Links ]
59. Montrasio F, Frigg R, Glatzel M, Klein MA, Mackay F, Aguzzi A, et al. Impaired prion replication in spleens of mice lacking functional dendritic cells. Science 2000;288:1257-1259. [ Links ]
60. Sy M-S, Gambetti P. Prion replication-once again blaming the dendritic. Nat Med 1999;5:1235-1237. [ Links ]
61. Blättler T, Brandner S, Raeber AJ, Klein MA, Voigtländer T, Weissmann C, et al. PrP-expressing tissue required for tranfer of scrapie infectivity from splen to brain. Nature 1997;389:69-72. [ Links ]
62. Brown P. The risk of bovine spongiform encephalopathy ("mad cow disease") to human health. JAMA 1997;278:1008-1011. [ Links ]
63. Hill AF, Desbruslais M, Jonier S, Sidle KCL, Gowland I, Collinge J, et al. The same prion strain causes vCJD and BSE. Nature 1997;389:448-450. [ Links ]
64. Zeidler M, Stewart GE, Barraclough CR, Bateman DE, Bates D, Burn DJ, et al. New variant Creutzfeldt-Jakob disease: Neurological features and diagnostic test. Lancet 1997;350:903-907. [ Links ]
65. Zeidler M, Johnstone EC, Bamber RWK, Dickens CM, Fisher CJ, Francis AF, et al. New variant Creutzfeldt-Jakob disease: Psychiatric features. Lancet 1997;350:908-910. [ Links ]
66. Will RG, Ironside JW, Zeidler M, Cousens SN, Estiberio K, Alperovith A. New variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996;347:921-925. [ Links ]
67. Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000;37:1-9. [ Links ]
68. Almond J, Pattison J. Human BSE. Nature 1997;389:437-438. [ Links ]
69. Aldhous P, Abbott A. Battling the killer proteins. Nature 2000;408:902-903. [ Links ]
70. Brown P, Preece M, Brandel J-P, Sato T, McShane L, Zerr I, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000;55:1075-1081. [ Links ]
71. Sardas RHK, Wilesmith JW. Bovine spongiform encephalopathy: Epidemiology, low dose exposure and risks. Ann NY Acad Sci 1994;724:210-220. [ Links ]
72. Schiermeier Q. Testing times for BSE. Nature 2001;409:658-659. [ Links ]
73. Organización Mundial de Sanidad Animal. Situación Sanitaria de la EEB. Office International des Epizzoties 2001. http//www.oie.int/esp/info/es_esbmonde.htm. [ Links ]
74. Thompson C. In search of a cure for CJD. Nature 2001; 409:660-661. [ Links ]
75. Reilly CE. ß-sheet breaker peptides reverse conformation of pathogenic prion proteins [letter]. J Neurol 2000;247:319-320. [ Links ]
76. Soto C, Kascsak RJ, Saborío CP, Aucouturier P, Wisniewski T, Prelli F, et al. Reversion of prion protein conformational changes by synthetic ß-shet breaker peptides. Lancet 2000;355:192-197. [ Links ]
77. Chabry J, Caughey B, Chesebro B. Specific inhibition of in vitro formation of protease-resistant prion protein by synthetic peptides. J Biol Chem 1998;273:13203-13207. [ Links ]
78. Caughey B, Race RE. Potent inhibition of scrapie-associated PrP accumulation by Congo red. J Neurochem 1992;59:768-771. [ Links ]
79. Farquhar C, Dickinson A, Bruce M. Prophylactic of pentosan polysulphate in transmissible spongiform encephalopathies [letter]. Lancet 1999;353:117. [ Links ]
80. Magné A, Nishida N, Milhavet O, McMahon HEM, Casanova D, Lehmann S. Amphotericin B inhibits the generation of the scrapie isoform of the prion protein in infected cultures. J Virol 2000;74:3135-3140. [ Links ]
81. Pocchiari M, Casaccia P, Ladogana A. Amphotericin B: A novel class of antiscrapie drugs. J Infect Dis 1989;160:795-802. [ Links ]
82. Adjou KT, Demaimay R, Lasmezas C, Deslys J-P, Seman M, Dormont D. MS-8209, a new amphotericin B derivative, provides enhanced efficacy in delaying hamsters scrapie. Antimicrob Agents Chemother 1995;39:2810-2812. [ Links ]
83. Tagliavini F, McArthur RA, Canciani B, Giaccone G, Porro M, Bugiani M, et al. Effectiveness of anthracycline against experimental prion disease in Syrian hamsters. Science 1997;276:1119-1122. [ Links ]
84. Priola SA, Caughey B, Caughey WS. Novel therapeutic uses for prophyrins and phthalocyanines in the transmissible spongiform encephalopathies [commentary]. Curr Opin Microbiol 1999;2:563-566. [ Links ]
85. Caughey WS, Raymond LD, Horiuchi M, Caughey B. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc Natl Acad Sci USA 1998;95:12117-12122. [ Links ]
86. Supattapone S, Nguyen H-O, Cohen FE, Prusiner SB, Scott MR. Elimination of prions by branched polyamines and implications for therapeutics. Proc Natl Acad Sci USA 1999;96:14529-14534. [ Links ]
87. Doh-ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 2000;74:4894-4897. [ Links ]
88. Faoro A, Filomena MV, de Pernía E, Borges J. Importancia del EEG en el diagnóstico de la enfermedad de Jakob-Creutzfeldt. A propósito de dos casos. Rev Ven Neurol Neurocir 1978;1:17-20. [ Links ]
89. Caraballo AJ. Creutzfeldt-Jakob disease in Venezuela: A case report. Arq Neuro-Psiquiat (Sâo Paulo) 1991;49:218-221. [ Links ]
90. Méndez O, Luzardo G, Molina O, Cardozo J. Enfermedad de Creutzfeldt-Jakob. Reporte de 2 casos. Invest Clín 1995;36:23-30. [ Links ]
91. Hernández A, Céspedes G, González A, Alvárez A, Lara C, Soto A, et al. Enfermedad de Creutzfeldt-Jakob en Venezuela: informe de 10 casos y revisión de la literatura. Rev Inst Nac Hig "Rafael Rangel" 1999;30:14-20. [ Links ]
92. Caruzo G, Díaz F, Bohórquez M, Rodríguez L, Gambetti P, Molina O, et al. Encefalopatía priónica familiar. XLIII Jornadas Venezolanas de Anatomía Patológica; nov 18-20; San Cristóbal, Venezuela. 1999.p.17. [ Links ]
Lista de Sociedades Afiliadas a la Red de Sociedades Científicas Médicas de Venezuela
Sociedad Venezolana de Anatomía Patológica
Dirección:
Calle López Aveledo, Edif. Centro Profesional Plaza, Piso 8, Ofic. 6-A Maracay – Edo. Aragua.
Sociedad Venezolana de Anestesiología
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos, PB, Ofc. 1-A. Urb. Santa Fe Norte.
Sociedad Venezolana de Alergia e Inmunología
Dirección:
S/D.
Sociedad Venezolana de Cardiología
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos, Piso 2, Ofc. B-1, Urb. Santa Fe Norte.
Sociedad Venezolana de Cirugía de la Mano
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos, Piso 2, Ofc. B-1. Urb. Santa Fe Norte.
Sociedad Venezolana de Cirugía Plástica, Reconstructiva, Estética y MF
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos, Piso 2, Ofc. B-1 Urb. Santa Fe Norte.
Sociedad Venezolana de Cirugía Ortopédica y Traumatología
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos , Piso 2, Ofc. B-1, Urb. Santa Fe Norte.
Sociedad Venezolana de Dermatología
Dirección:
Avda. José Ma. Vargas. Torre Colegio de Médicos, Piso 2, Ofc B-1, Urb. Santa Fe Norte.
Sociedad Venezolana de Endocrinología
Dirección:
Avda. El Golf. Qta. Setenta y seis, Urb. El Bosque.
Sociedad Venezolana de Geriatría y Gerontología
Dirección:
Calle Caurimare cruce con Caroní, Edf. El Carmen Apto. A-5, Colinas de Bello Monte.
Sociedad Venezolana de Infectología
Dirección:
Avda. José Ma. Vargas, Torre Colegio de Médicos, piso 2 Ofic. 2-E Urb. Santa Fe Norte.
Sociedad Venezolana de Medicina Crítica
Dirección:
Avda. El Golf. Qta. Setenta y seis, Urb. El Bosque.
Sociedad Venezolana de Medicina del Deporte
Dirección:
Avda. Principal Santa Fe Sur, Resd. Araucaria Nº 7428*AH