










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica de Caracas
versión impresa ISSN 0367-4762
Gac Méd Caracas v.113 n.1 Caracas ene. 2005
La recuperación del miocardio hibernado mejora el pronóstico de la cardiopatía isquémica metabólica
Dr. Fernando Bermúdez Arias*
*Cardiólogo. Investigador. Universidad del Zulia. Presidente de la Asociación Venezolana de Aterosclerosis
Trabajo de incorporación como Miembro Correspondiente Nacional Puesto Nº 13. Presentado en la Academia Nacional de Medicina en la sesión del 20 de noviembre de 2003.
RESUMEN
El significado del aturdimiento y de la hibernación del miocardio, los mecanismos fisiopatológicos responsables de estos fenómenos y las posibles conexiones entre ambos han sido objeto recientemente de múltiples revisiones. El aturdimiento es la disfunción contráctil de carácter transitorio y duración variable (minutos a semanas), que sobrepasa el período de isquemia aguda o subaguda. Sin embargo, su trascendencia es limitada, porque responde muy bien a la estimulación inotrópica y se resuelve progresivamente de forma espontánea. Sus mecanismos fisiopatológicos no son bien conocidos, y todavía se debate si se debe fundamentalmente a las secuelas de la lesión isquémica o a un efecto colateral indeseable de la reperfusión, especialmente en la fase inicial de esta, durante la que se producen radicales libres de oxígeno cuando se hace en forma rápida (cirugía o angioplastia), lo cual no sucede cuando se logra en forma lenta (tratamiento medicamentoso).
El miocardio aturdido e hibernado se define como una alteración persistente de la función miocárdica del ventrículo izquierdo en reposo, ocasionada por una reducción del flujo miocárdico.
Pero la definición de la hibernación es controvertida, debido a que no se conoce el mecanismo de la disfunción cardíaca aparentemente crónica, dependiente de una cardiopatía isquémica, la cual puede desaparecer después de la revascularización. La respuesta hibernante del corazón fue considerada como un acto de auto preservación (poco flujo poco trabajo), considerándose así como el "corazón inteligente". Se ha demostrado que la disminución severa del flujo coronario puede dar lugar a una reducción aparentemente adaptativa de la actividad contráctil y de otras funciones, de horas, días, meses y años de duración, sin muerte celular, pero con mantenimiento de los niveles de adenosintrifosfato. Igualmente, en algunos casos de hibernación crónica el flujo coronario puede ser normal en reposo, y se ha sugerido que esta situación podría explicarse por episodios repetitivos y predominantemente silentes de isquemia-aturdimiento-hibernación. También Interviene en ese sentido el desarrollo de la circulación colateral, dependiente del precondicionamiento isquémico del miocardio, que se define como el incremento de tolerancia a la isquemia prolongada proporcionado por uno o más ciclos de isquemia breve y reperfusión
La estabilización del trombo por fibrosis, que anula la imposibilidad de disgregarlo, el estado metabólico del miocardio, el grado inflamatorio, determinado por la presencia de polimorfonucleares y la extensión del área infartada inicialmente, así como el predominio del miocardio necrosado y fibrosado sobre el hibernado, hacen más difícil la recuperación del miocardio hibernado a la normalidad. La diferenciación del miocardio hibernado (viable) del miocardio no viable en un paciente con cardiopatía isquémica metabólica y disfunción ventricular izquierda es un punto clave en la era actual de la revascularización miocárdica.
En esta investigación clínica de 12 años de duración, en 7 049 casos de cardiopatía isquémica metabólica aguda, subaguda y crónica, sin inclusión selectiva ni exclusión de casos, demostramos que en todos ellos hay miocardio hibernado, el cual puede ser recuperado con tratamiento médico dietético farmacológico causal, y mantenidos asintomáticos con electrocardiograma recuperado en proporción aproximada entre el 90 % al 95 %.
SUMMARY
The physiopathologycal mechanisms of both stunning and hibernating myocardium and the connections between them have recently been the objet of multiple revisions. Stunning is the contractil disfuntion of transitory character and variable duration (minutes to weeks), as consecuense of an acute or subacute ischemia period. Nevertheless, is of limited importance because there is a good response to the inotropic stimulation and it is progressively solved in a spontaneous manner. Their physiopathological mechanisms are not known and it is still debated whether it is fundamentally an ischemical sequel damage or a reperfusion undesirable collateral effect. This is especially true in the reperfusion initial phase when a fast oxygen free radicals increase takes place (either by coronary bypass surgery or by percutaneous transluminal coronary angioplasty). This does not occur when it is slowly done (pharmacologic revascularization).
Stunning and hibernating myocardium is defined as persistently impaired myocardial and left ventricular function at rest, resulting from reduced myocardial blood flow. It is postulated that despite the reduced coronary blood flow, metabolic activity is sufficient to prevent tissue necrosis. The hibernating myocardial definition is controverted, as the heart chronic disfunction in not well known because the it is coronary artery disease dependent. The heart hibernating response namely a cardiac function reduction to cope with a reduced myocardial blood flow, has been considered a self preservation act (little blood, little work) for which the hibernating heart is thought of "a smart heart". It have been show that a coronary flow severe decrease can give rise to an adaptive contractile activity and other heart functions reduction, within hours, days, months or years of duraction, without cellular death, but with adenosintriphosphate maintenance levels. In some cases of chronic hibernation, the coronary flow can be normal at the rest, suggesting that this situation could be explained by itself as repetitive and predominantly ischemia-stunning-hibernating silent episodes. Also, collateral circulation development takes palce, dependent on the ischemic preconditioning myocardium. This is defined as the tolerance increase to prolonged isquemia provided by one or more short of isquemia cycles followed by reperfution.
The thrombus stability by fibrosis wich avoids its disintegration possibility, the myocardial metabolic state, the inflammatory process degree (determined by the polimorphonuclears presence) the previous necrotic area extention and the necrotic and fibrous myocardium predominance over the hibernating one, make more difficult the hibernating myocardium recovery towards normality. The hibernating myocardium differentiation from nonviable myocardium in patients with coronary artery disease and left ventricular dysfunction is a key issue in the current myocardial revascularization.
In this 12 years long clinical investigation of 7 049 cases with acute, subacute and chronic metabolic heart disease, neither with selective inclusion nor exclusion of cases, we showed than there was hibernating myocardium in all of them, which was recovered (asyntomatic) with pharmacological-dietetic-causal treatment with near-normal electrocardiogram, recovered in a proportion around 90 % to 95 %.
INTRODUCCIÓN
La cardiopatía isquémica metabólica (CIM) por aterotrombosis coronaria es la enfermedad más frecuente y de mayor mortalidad del siglo veinte, a tal punto que se considera como la epidemia de este siglo. La padecen en Estados Unidos (1995) 1,5 millones de personas al año, de las cuales 300 000 mueren antes de llegar al hospital y 200 000 al mes de haber ocurrido el infarto del miocardio. El porcentaje de mortalidad que ocasiona en la población general es del 33 % en el primer mes y del 10 % en el primer año (1). En Venezuela, de 67,7 muertes por cien mil habitantes por año, en 1986, subió a 84,1 en 1990 (2). En España, en 1994 fue de 98 en hombres y de 60 en mujeres por cada 100 000 habitantes ajustados a la edad por año (3). De acuerdo al estudio MRFIT (1) la relación de mortalidad con colesterol sérico de 260 mg/dL o más fue de 1 200 por cada 1 000 habitantes, y según el estudio de Framingham a los 10 de años de observación continua los varones no diabéticos murieron aproximadamente 1 000 por cada 100 000, mientras que los diabéticos duplicaron la cifra. En mujeres las cifras fueron de 400 y 2 000 respectivamente (4). En este trabajo expondremos el concepto derivado del razonamiento de un tratamiento dietético farmacológico causal, enfocado desde el punto de vista de la disgregación del trombo y de la recuperación del tejido hibernado y sus resultados.
Definición y fisiopatología
La principal causa de este trastorno isquémico-metabólico cardíaco es la trombo aterosclerosis coronaria, base de la trombosis plaquetaria y su consecutiva obstrucción arterial coronaria, acelerada por el estrés, la diabetes, el mixedema, las dislipidemias, la disminución de la lipoproteinemia de alta densidad, el tabaco, el alcohol, las drogas psicotrópicas, el sedentarismo, la hipertensión, la hipertrofia ventricular izquierda (HVI) y otros factores de riesgo conocidos o no, conjunto que produce la enfermedad isquémica metabólica del corazón bajo cualquiera de sus manifestaciones (angina de pecho, infarto del miocardio, arritmias, insuficiencia cardíaca y muerte (súbita o no). La aterosclerosis es la causa subyacente más común, pero es la ruptura de la placa, justamente en el sitio donde ésta se une al endotelio sano, el lugar habitual donde sucede, por ser el sitio más frecuente donde se descubre el colágeno, para dar inicio a la cascada trombótica (adhesión y agregación plaquetaria) y a la producción de tromboplastina tisular, sustancia que desencadena la cascada de la coagulación por la vía extrínseca. De esta manera, la aterosclerosis es el factor subyacente y la trombosis así producida la causa principal de la oclusión aguda de las arterias coronarias. El mecanismo inmunológico que transforma y perpetúa la lipoproteína de baja densidad (LDL)-colesterol en LDL oxidada, es posiblemente el factor más importante en la producción de la aterosclerosis (5-10).
Se sabe que el proceso aterosclerótico inicial se debe al exceso de LDL, de carga positiva, que al no poder ser atrapado por los receptores de los tejidos, penetran a la íntima arterial (espacio subendotelial) donde se radican estas moléculas por efecto electrostático de los proteoglicanos, de carga negativa, en el proceso que se ha denominado "anclaje". Luego ocurre la oxidación de esas LDL, primero por alteración de sus puentes disulfuros y más adelante por englobamiento de los macrófagos (MCF), que son atraídos por ellas. Una vez englobados se producen radicales libres de oxígeno (RLO) en el interior del macrófago, mediante los cuales el MCF intenta destruir las LDL. Al no lograrlo y al no haber autolimitación de llenado de LDL, el proceso continúa, formándose así las llamadas células espumosas, manifestación primaria de la aterosclerosis, y al mismo tiempo, mediante procesos inmunológicos, ocurre también que las células musculares lisas (CML), por efecto quimiotáctico también se hacen presentes en la íntima arterial, sitio donde ceden tejido conectivo que engloba las células espumosas, sufriendo ellas también el proceso que las conduce a células espumosas. De este modo se conforma una pequeña placa fibrosa localizada en la íntima o una estría ("estría grasa"), la cual ya puede asomarse a la luz del vaso. De cualquier manera llega el momento que ellas se llenan tanto de LDL-ox, que explotan y rompen el endotelio, pero por estar muy cerca de la luz del vaso, liberan esas moléculas de LDL-ox a la sangre, aumentando así su concentración. Rápidamente el sitio ocupado por la placa rota se reendoteliza, dando posteriormente un período de calma, cuya duración depende de la agresividad de los factores que lo han conformado, y es el proceso inflamatorio el predominante en el acortamiento de esa acalmia. De la intensidad de los factores de riesgo la placa puede progresar y agrandarse, pero también puede suceder que en vez de reendotelizarse, sufra el proceso de la trombosis plaquetaria, ocluyendo la arteria, conduciendo de esta manera al infarto del miocardio o a la angina inestable. La placa en sí misma, sin importar su tamaño, es muy sensible a la ruptura, especialmente en su unión con el endotelio sano. Antes que suceda esta manifestación morfológica de la enfermedad, ya se presentan alteraciones funcionales del endotelio (disfunción endotelial) como consecuencia de la placa que ya está formada, que se manifiesta por disminución de la vasodilatación, debido a una disminución en la producción del factor de relajación endotelial (óxido nítrico) (11,12).
Del mecanismo aterotrombótico mencionado se desprende la importancia de combatir el estado proinflamatorio general y estabilizar y regresar la placa aterosclerótica con dieta hipolipemiante (13) y medicamentos antilipídicos (14), antioxidantes (15-20) y promotores del aumento del poder inmunológico, como se ha demostrado recientemente con la vacuna antiinfluenza (20). Pero así mismo, hay otras causas que provocan algunas de sus manifestaciones, como por ejemplo, la HVI, la crisis de hipertensión arterial, la miocardiopatía hipertrófica obstructiva, la estenosis aórtica, en las cuales puede presentarse angina de pecho por insuficiente riego arterial debido a la desproporción entre la fibra hipertrofiada y el alcance de la arteriola, o al desequilibrio entre la oferta y la demanda, al aumentar esta última. Igualmente, la angina por enfermedades pulmonares y la hipertensión pulmonar debido a la hipoxia; la angina en la policitemia vera y otras causas de hiperhemoglobinemia (uso continuo y a largo plazo de vitamina B12 y protectores hepáticos), que aumenta la viscosidad sanguínea y disminuye la velocidad circulatoria; la angina de pecho por otras causas como las arteritis por colagenopatías, la enfermedad de Takayashu y la enfermedad de Leo Burger, y por último, la que es consecuencia de la anemia, de la disminución mantenida del llenado diastólico coronario, como se observa en las arritmias paroxísticas con frecuencia ventricular elevada y la que se debe a isquemia funcional, como es la evidencia de algunos casos de prolapso de válvula mitral (PVM), posiblemente ocasionada por la tracción sufrida de los músculos papilares como consecuencia del estiramiento de los pilares por la excursión hacia atrás de los velos de la mitral y también los observados en el síndrome X. En el PVM, la isquemia subepicárdica suele aparecer en D2, D3 y aVF (Barlow), o en V1, V2, V3 y a veces hasta V4 (Bermúdez Arias). La T negativa infantil, que es el diagnóstico diferencial en la última manifestación mencionada, por lo general no pasa de V2, y tiene morfología diferente (menos/más), y no negativa primaria como en el PVM de la valva anteroseptal, como fue descrita por nosotros (15).
Toma importancia por tanto, la interrelación de los mecanismos de producción que conducen a la CIM (15-23), como son la aterosclerosis, la enfermedad hipertensiva esencial (EHE), la diabetes, la dislipidemia y la obesidad, y también aquellos que ocasionan un menor aprovechamiento (alimentación inadecuada, sedentarismo) o una menor utilización de los principios inmediatos, entre los cuales figuran la oxidación, y en el área cardíaca, el atontamiento y la hibernación (22,23). La representación gráfica de la interrelación de estos mecanismos de producción bajo la concepción de una intersección compleja, con la finalidad de aproximarnos al pronóstico de la CIM, la concebimos en 1981 mediante la figura geométrica denominada "intersecancia", término cuyo significado fue discutido y aceptado en los Congresos Mundiales de Matemáticas, en Bogotá (1980), y en México (1990) (Fernández Mario, profesor de Filosofía y Enseñanza de la Matemáticas de la Universidad del Zulia) (Figura 1). En efecto, entre los tres mecanismos fisiopatológicos primarios fundamentales y causales de la CIM: A1. Menor aporte sanguíneo por aterosclerosis coronaria. A2. Deficiente utilización de los principios inmediatos (diabetes, oxidación, antioxidación por acción de los radicales libres de oxígeno, atontamiento, hibernación). A3. Aumento de la demanda por EHE con HVI, se deduce que si cada uno de ellos se expresa por círculos parcialmente superpuestos, se originan tres nuevas secciones comunes y críticas (B1, B2, B3), las cuales, comparadas con las originales, representan un peor pronóstico, y una sección central donde confluyen los tres mecanismos primarios (C), que a su vez constituye la situación más grave (17). Cuando, como ha sido descrito, esos mecanismos se intersecan, da como resultado la figura geométrica que se denomina en matemática aplicada intersecancia, la cual empleada en la medicina nos permitió en aquella época (1981) valorar el pronóstico de la cardiopatía isquémica en lo que designamos "Un nuevo modelo de comprensión de la cardiopatía isquémica" (17), y que al mismo tiempo fue la base que nos condujo al entendimiento y desarrollo del nuevo concepto del miocardio hibernado, que para entonces se iniciaba (Diamond y col., 1978) (22), así como a la iniciación del tratamiento y recuperación de dicho miocardio, desarrollado por nosotros en los siguientes años, y cuyos fundamentos diagnósticos, terapéuticos y resultados exponemos en este trabajo.
La utilización en medicina de la figura geométrica "intersecancia" orienta el valor pronóstico de la enfermedad isquémica cardíaca. Así, de acuerdo a las observaciones clínicas, cuando dos o más mecanismos de producción primarios (Sistemas cerrados o círculos en este caso), se intersecan y forman secciones de intersección comunes, aunque críticas, diferentes a las originales, se concluye que éstas, en sí mismas, representan una mayor gravedad de dicha enfermedad.
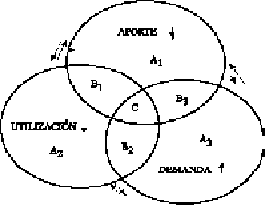
| A1: menor aporte Aterosclerosis coronaria A2: menor utilización Diabetes Oxidación > antioxidación Miocardio atontado Miocardio hibernado A3: mayor demanda Enfermedad hipertensiva esencial Hipertrofia ventricular izquierda | Empeoramiento del pronóstico Sección B1 > A1 y A2 Sección B2 > A2 y A3 Sección B3 > A1 y A3 Sección C > A1, A2, A3, B1, B2, B3 |
|
|
|
Bermúdez Arias F (17)
Figura 1. Nuevo modelo de compresión de la cardiopatía isquémica (1981) (Modificado, 2003)
A continuación se explican más ampliamente los mecanismos primarios y se razona fisiológicamente su producto final: CIM: 1) Disminución del aporte. La causa más frecuente de este mecanismo es la tromboaterosclerosis coronaria, enfermedad multifactorial, entre cuyos factores de riesgo más importantes se encuentran la dislipidemia (aumento de colesterol por encima de 150 mg/dL, de LDL por encima de 100 mg/dL de triacilglicéridos, por encima de 120 mg/dL y disminución de lipoproteína de alta densidad (HDL) por debajo de 45 mg/dL, hipertensión arterial, sedentarismo, tabaquismo, obesidad, diabetes miellitus, resistencia a la insulina, y siguiéndole el espasmo de las arterias coronarias, el síndrome X coronario, los puentes musculares, los obstáculos en el ostium de estas arterias, como sucede, por ejemplo, en la sífilis, las embolias, bien originadas en las cámaras izquierdas del corazón o en las propias arterias (aneurisma micótico en la endocarditis bacteriana subaguda) y el aumento de grosor de la fibra miocárdica, sea por hipertrofia secundaria a la hipertensión o al entrenamiento físico o por la presencia de una miocardiopatía hipertrófica. También el PVM, como ya se analizó. Estas causas disminuyen o anulan totalmente el flujo de una arteria hacia el miocardio, ocasionando un desequilibrio entre la oferta y la demanda requerida por dicho tejido. 2) El segundo factor, el cual trabaja en el mismo sentido, es decir, desequilibrando la oferta y la demanda, pero ahora aumentando esta última, como acontece en la hipertensión arterial sistémica, especialmente cuando se acompaña de hipertrofia ventricular izquierda. 3) El tercer mecanismo fisiológico es el metabólico, como el que se observa en la diabetes, por ejemplo, en el cual disminuye el aprovechamiento energético de la fibra con la consiguiente disminución de su capacidad bioquímica y de su efecto mecánico. En el mismo sentido metabólico actúa la hipokalemia. Últimamente también se ha observado que el efecto deletéreo metabólico más importante lo producen los RLO, al destruir la membrana celular y mitocondrial, permitir la salida de K+ y la entrada exagerada de Na+ y Ca++, alteraciones que no solamente complican a la isquemia, sino que crean otras nuevas más agresivas y perpetuadoras del proceso fisiopatológico, como se verá más adelante. Asimismo, la disminución de adenosintrifosfato (ATP), especialmente cuando ya hay isquemia y se reperfunde el miocardio isquémico en forma rápida.
Aunque la causa principal de la CIM es la tromboaterosclerosis, sin embargo, cualquiera que sea el mecanismo de la isquemia, la alteración fisiopatológica inicial, caracterizada por disminución de la producción de ATP a partir del oxígeno, siempre se acompaña de otras manifestaciones fisiopatológicas de origen metabólico, que tienen su causa en la producción de RLO, los cuales aumentan cuando los metabolitos intermedios para llegar al ATP, como son adenosindifosfato (ADP), adenosimonofosfato (AMP), isoxantina, xantina y adenosina, mediante la acción de enzima xantino oxidasa producen mayor cantidad de RLO. Estas sustancias, de acción muy breve, actúan sobre los lípidos de la membrana, provocando destrucción de ésta, y consecutivamente trastornos de los electrolitos (salida de K+ y entrada de Na+ y Ca++), con menor poder energético y hasta muerte celular. De esta aseveración se ha concluido en denominar a esta enfermedad "CIM" y no cardiopatía isquémica o insuficiencia coronaria, como se le conocía hasta el presente (13). No obstante, de las mencionadas alteraciones establecidas crónicamente, surge un mecanismo extraordinario de defensa: el miocardio hibernado, motivo de esta investigación clínica.
MATERIAL Y MÉTODO (Cuadros 1 a 3)
Nuestras observaciones clínicas y electrocardiográficas en humanos fueron demostradas inicialmente en dos trabajos comparativos (18,19) y posteriormente en uno más, este último abierto y mantenido bajo los mismos principios y con el mismo propósito de revascularizar farmacológicamente, y recuperar el miocardio hibernado, desde 1986 hasta 1998, motivo de esta comunicación (17-21).
Cardiopatía isquémica metabólica (CIM)
| Enfermedades asociadas ( % ) |
|
Cardiopatía isquémica metabólica (CIM)
| Estudios ( % ) |
|
Tratamiento médico dietético farmacológico casual, estado basal y distribución de los casos
7 049 casos sucesivos en 12 años de seguimiento, sin selección ni exclusión de casos. Estudio abierto no comparativo (1984-1996)
| Femenino (%) | Masculino % | 30-66 años % | 66-80 años % | Mas de 80 años % |
| 67 | 33 | 6 | 74 | 20
|
CIM aguda % CIM subaguda % CIM crónica %
________________________________________________________________
8 25 67
________________________________________________________________
Tratamiento (Cuadro 4)
Se basa en que la mayoría de los trastornos cardiológicos ocasionados por la CIM se deben en buena parte y afortunadamente a la presencia de tejido hibernado inmerso dentro del tejido fibrosado (aparentemente necrosado en su totalidad o alterado parcialmente en su estructura). Por otra parte, como el tejido miocárdico hibernado es viable en cualquier momento de su evolución siempre que se logre revascularizar y proteger al mismo tiempo otras complicaciones como la presencia de RLO y de la ruta metabólica predominante por vía de los ácidos grasos, concluimos que de lograrlo mediante el tratamiento médico dietético farmacológico causal se puede lograr reactivar total o parcialmente (esto último con mayor probabilidad) el potencial de acción transmembrana (PAT) y el potencial de recuperación transmembrana (PRT) de los miocitos hasta entonces disminuidos y en consecuencia reactivar las funciones eléctricas y mecánicas del corazón. Las bases racionales del tratamiento médico (no intervencionista ni quirúrgico) se basan en el tratamiento racional (causal) de la cardiopatía isquémica metabólica al revascularizar lentamente el miocardio, pero que al mismo tiempo tenga el propósito de recuperar el miocardio hibernado, bajo los siguientes criterios: 1. Aumento de la molécula de ATP, mediante la reperfusión lenta por disgregación del trombo plaquetario, consecutivo a la ruptura de la placa aterosclerótica (al mismo tiempo que desarrollando circulación colateral con el ejercicio) y mediante el ahorro de esas moléculas al cambiar la ruta metabólica de ácidos grasos a carbohidratos con citoprotectores y con el aporte alimentario gracias a la dieta adecuada (fisiológica, cronobiológica, antioxidante, polarizante) (16), al mismo tiempo que evitando el consumo inútil del ATP (estrés). 2. Evitando la oxidación mediante la dieta adecuada (evitar alimentos procesados), disminuyendo su producción consecutiva a la reperfusión rápida y combatiendo las enfermedades oxidativas como la diabetes, a la vez que combatiendo la producción de RLO con citoprotectores. 3. Manteniendo el potasio en cifras no inferiores a 4,8 mEq/L en plasma, confirmando su estado intracelular mediante el ECG-12D. Su aporte principal se logra con la dieta polarizante y cuando está muy bajo, con solución polarizante y soporte farmacológico oral. 4. Disminuyendo el estado inflamatorio arterial y miocárdico y estabilizando la placa (estatinas, fibratos, vacuna antiinfluenza).
Esquema de tratamiento de acuerdo al tipo de CIM y con el propósito de recuperar el miocardio hibernado
| Concepto | Clase/sust.act | Acción | Medicamento | Dosis |
| CIM aguda | 1) Fibrinolítico estreptokinasa | Destruye fibrina | * Kabikinase * Estreptase | 1,5 M de U |
|
| 2) HBPM Dalteparina Enoxoparina Nadroparina | -IIa -Xa - AGR. Plaq. Fibrina | * Fragmin * Clexane * Fraxiparina | 5 000 UI D X 10d 4 000 UI 3 000 UI |
|
| 3) Anti agreg. plaq. Indobufeno Ticlopidina Clopidogrei | - Recent TXA2 -ADP - ADP | *Ibustrin * Ticlopidina * Plavix | 660 mg/d 500 mg/d 75 mg/d |
| 4) Citoprotector/anti-RLO | ||||
|
| Trimetazidina | Citoprotect. Inhibe RLO | * Vastarel | 60 mg/d |
|
| 5) Potasio Gloconato de potasio | Mejora hipo K | Sol. polarizante | 60 mEq/d |
|
| 6) Antigonist del Ca Amlodipina Nifepidina Diltiazem Verapamil | Disminuye canal lento de calcio | * Norvas * Adalat * Diltiazem. * Tilazem * Manidon | 5 mg/d 30 mg/d 900 mg 240 mg/d |
|
| 7) Misceláneos | Fibrinógeno | Frental | 400 a 1 200 mg/4 |
|
| Pentoxilina | Ciprofibrato | Hiperlipen | 100 mg/d |
|
| 8) Antioxidantes Vit. E Vit. C Gimko biloba | LDL oxidada | Ephinal Cebion Kiadon, Tanakan | 400 a 600 U/d 1 g/d 120 mg/d |
|
| 9) Estatinas, fibratos | Hipolipemian | Atorva, Simva, Prava, ROSU | 10-40 mg/d |
|
| 10) Dieta F-Cap | Fisilógica /cronobiológica/ antioxidante/ Polarizante |
|
|
|
| 11) Ejercicios Físicos | Precondionam. Adenosina Circulación colar. |
|
|
| CIM sudagud |
2+3+4+5+6+7+8+9 |
|
|
|
| CIM crónica | 3+4+5+6+7+8+9+10 |
|
|
|
Bermúdez Arias, 1990 Electrocardiografía diagnóstica, McGraw-Hill Interamericana de Venezuela.
El conjunto del tratamiento lo he resumido en la figura del árbol de la cardiopatía isquémica (20), el cual se alimenta con los factores de riesgo (aproximadamente 36), habiendo concluido que los tratamientos actuales (convencionales, incluyendo el médico: aspirina, betabloqueantes, diuréticos y vasodilatadores coronarios de duración prolongada), la angioplastia con stent o sin éste, y la cirugía, sólo atacan el ramaje de ese árbol, cuya conclusión es la permanencia de la enfermedad aterosclerótica, no solamente en el corazón, sino también en el cerebro, riñón, carótidas, aorta, mesenterio, retina y miembros inferiores. Para acabar con el árbol es necesario arrancar la raíz, que sería como acabar con la aterosclerosis, lo cual no se vislumbra en la actualidad, o cortar el tronco, que es lo que se propone con este tratamiento, al disgregar el trombo (cualquiera que sea el tiempo de constituido), proteger de los RLO (antioxidar), mantener el potasio y recuperar y mantener el ATP en su máxima expresión. Los medicamentos utilizados, como antiagregantes plaquetarios (heparina de bajo peso molecular, indobufeno y clopidogrel), cardioprotectores (trimetazidina y potasio) y estabilizadores de la aterosclerosis (estatinas, fibratos y vacuna antiinfluenza) se resumen en el Cuadro 4.
RESULTADOS
Del tratamiento realizado, hemos concluido que la zona inactivable no representa únicamente tejido muerto, sino que es una mezcla de tejidos muertos, aturdido e hibernado, ya que mediante la "trombectomía química" lograda con el tratamiento dietético farmacológico utilizado para tal fin y que analizaremos más adelante, hemos observado en aproximadamente el 97 % desaparición de los síntomas anginosos, por lo general antes del tercer día de iniciado el tratamiento, posiblemente debido a la rápida desaparición del ácido láctico al activarse la creatinfosfoquinasa; en el 92 % normalización de las ondas de lesión y de las T invertidas y en el 34 % disminución o desaparición de la onda Q patológica, todo ello en 7 049 pacientes con CIM (aguda, subaguda y crónica) observados durante doce años, sin que hubiese selección ni exclusión durante ese período de tiempo.
Resultados generales (12 años). Tratamiento médico dietético farmacológico causal
|
| Primer período Primeros 6 años % | Segundo período Segundos 6 años % | Total 12 en 12 años % |
| Mortalidad | 0,12 | 0,19 | 0,31 |
| Reinfarto | 0,21 | 0,35 | 0,56 |
| Reanginas | 1,20 | 1,63 | 2,83 |
| Cirugía revascularización | 0 | 0,056 | 0,056 |
| Angioplastia | 0 | 0 | 0 |
| Mejoría o desaparición del dolor precordial | 56 | 41 | 97 |
| Mejoría de disnea cualquier grado | 58 | 38 | 96 |
| Frecuencia cardíaca de taq. sin(>90 por min) a FC normal (70-90 por min) |
50 |
40 |
90 |
| Frecuencia cardíaca De brad. Sin (>70 por min) a GF normal (50-90 por min) |
44 |
41 |
85 |
| Mejoría ECG de Lesión e isquemia | 50 | 42 | 92 |
| Mejoría de la zona eléctricamente inactivable | 23 | 10,4 | 33.4 |
* En el estudio de Framingham, partiendo de personas sanas no diabéticas a los 11 a 15 años, la mortalidad fue de 1,3 en hombres y 0,9 en mujeres mientras que en los diabéticos fue de 3 en ambos sexos. Obsérvese que nuestros resultados arrojan una cifra inferior (0,3 %) no obstante haberse iniciado el tratamiento cuando ya estaba instalada la CIM en Bermúdez Arias F 2002.
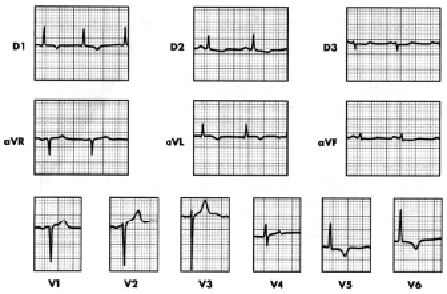
Figura 2. Hombre de 53 años, diabético con EHE. Criterios de HVI-OB de isquemia subepicárdica (T negativa primaria a en D1, VL, V5 y V6, y positiva en aVR). Además IM septal medio y bajo.
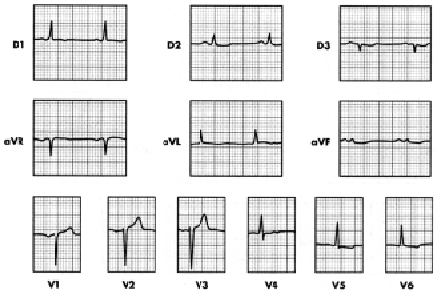
Figura 3. Paciente anterior. Después de 15 días de tratamiento hipotensor y antiisquémico se observa progresión de HVI-I, pero con disminución de la isquemia subepicárdica en D1, VL, V5 y V6. Onda T negativa en aVR (normal). IM septal sin modifiación.
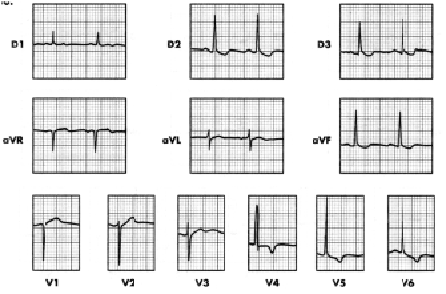
Figura 4. Mujer de 72 años. con EHE y diabética II. HVI-IIB (SS en V5, V6). Isquemia subepicárdica inferior (D2, D3, aVF) + septal baja y lateral baja. Onda T positiva en aVR.
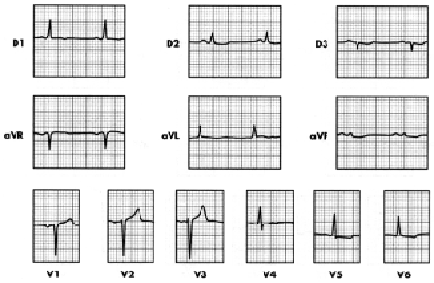
Figura 5. A los 15 días de tratamiento antihipertensivo y antiisquémico se observa regresión de la SS el VI y de la isquemia inferior, septal baja y lateral baja. T negativa en aVR.
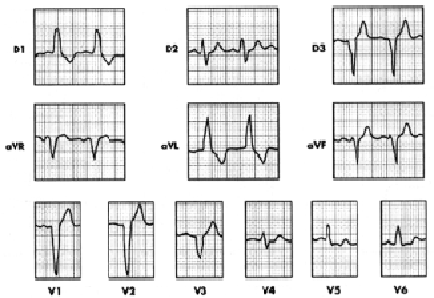
Figura 6. Hombre de 65 años, EHE. HVI-IVA (BCRIHH) + CIM + signos de ICC. IM septal bajo (RS en V4).
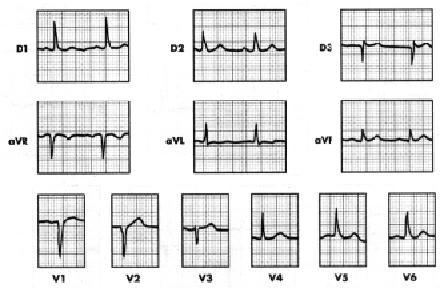
Figura 7. A los 6 meses: disminución de HVI (HVI-OB). Aparece R y T positiva normal en V3, que indican revascularización en esta área. Desaparece isquemia inferior. Q patológica en D3 (IM inferior, oculto en el ECG anterior por el BRI).
DISCUSIÓN (21-32)
La relación más intensa entre el flujo coronario, el consumo de oxígeno y el desempeño contráctil del corazón, es fundamento de la fisiología cardíaca, debido a la pequeña capacidad que tiene el miocardio de extraer oxígeno de sus reservas (mioglobina). Una reducción del flujo sanguíneo coronario, rápidamente disminuye el desempeño contráctil del miocardio (21), relación también investigada en perros sometidos a disminución progresiva del flujo coronario. Estos estudios han demostrado la existencia de una relación muy cercana, o mejor dicho, de un acoplamiento entre el suministro de substratos del miocardio (incluyendo oxígeno) y la demanda miocárdica de energía. La disminución proporcional entre el flujo miocárdico y su función contráctil ha sido llamada "acoplamiento agudo entre la perfusión y la contracción". La evolución natural de la cardiopatía isquémica metabólica incluye una serie de eventos que comienzan mucho tiempo antes de la aparición de las manifestaciones clínicas y electrocardiográficas que la caracterizan. Tal vez, el fenómeno inicial, aproximadamente 30 años antes es la pérdida progresiva de las funciones endoteliales normales que hoy se conoce como disfunción endotelial. Como consecuencia de ésta, finalmente se llega al desequilibrio: 1. Protrombótico/antitrombótico, el cual puede desembocar en un fenómeno aterotrombótico isquémico coronario agudo, subagudo o crónico. 2. Vasodilatación/vasoconstricción, cuyo mecanismo de producción agudo más frecuente es el vasoespasmo coronario, pero que también puede ser mantenido constituyendo entonces la EHE. Ambas alteraciones fisiológicas contribuyen a su vez a una mayor disfunción endotelial.
En el pasado reciente se creía que un fenómeno isquémico grave y prolongado, propio del infarto del miocardio, desembocaba necesariamente en la muerte de todo el tejido miocárdico sometido a esa alteración isquémica y metabólica, que tenía su expresión máxima en la presencia de la onda Q patológica del electrocardiograma, denominada zona inactivable o zona de necrosis. Actualmente, gracias a las observaciones de Diamond (22), en 1978, en pacientes con enfermedad arterial coronaria crónica, sin infarto del miocardio previo, pero con anormalidades crónicas de la motilidad de la pared ventricular izquierda, que se revertían con la revascularización quirúrgica, Rahimtoola (23) dedujo que gracias a una respuesta poco común del miocardio en esas condiciones, este tejido disminuía su requerimiento de oxígeno y llegaba a un nuevo estado de equilibrio, en el cual el miocardio no sufría necrosis ni el paciente sentía síntomas isquémicos. Esta última aseveración de Rahimtoola (23), en 1989, al estudiar los hallazgos de Diamond, implicaba según JLJ Vamoverschelde y col. (24) que: 1. El corazón puede adaptarse espontáneamente a un estado de hipoperfusión crónica, 2. El corazón llega a un nuevo estado de equilibrio entre la perfusión y la contracción. 3. Este nuevo equilibrio se puede mantener por un período prolongado de tiempo. 4. En nuestros resultados resaltamos el hecho que este tejido en estado de hipoperfusión crónica, pero viable (hibernado), es recuperable cuando se logra su reperfusión y se protege de los RLO, sin importar el tiempo que tenga en esas condiciones.
La reperfusión después de períodos cortos de isquemia (menos de diez minutos), usualmente concluye en una rápida y completa restauración de las funciones cardíacas, con mantenimiento normal de la ultraestructura miocárdica. Períodos más prolongados, entre quince y veinte minutos, usualmente no causan necrosis tisular, pero al efectuarse la reperfusión correspondiente a ésta se asocia una disfunción casi siempre reversible, que se denomina atontamiento o aturdimiento miocárdico (11,12), que se debe en gran parte al efecto deletéreo metabólico que ejercen los RLO sobre las biomoléculas de las células miocárdicas (25). Incrementos mayores en la duración de la isquemia usualmente resultan en grados variables de daño celular irreversible. La secuencia final de este proceso es lo que se observa clínica y electro cardiográficamente en el infarto del miocardio agudo, caracterizado en el ECG-12D por la presencia de lesión, isquemia y necrosis.
La hibernación miocárdica ocurre como resultado de una respuesta adaptativa a una reducción sostenida e importante del flujo miocárdico en reposo, tal como se observa en la isquemia miocárdica severa crónica, con o sin infarto del miocardio previo. Bajo condiciones agudas de isquemia, y por tanto, bajo el efecto del aturdimiento miocárdico, la contracción miocárdica se acopla de manera proporcional al suministro sanguíneo disponible para un momento dado. Esto ha inducido a los investigadores a examinar si un acoplamiento perfusión/contracción sostenido pudiese llevarse a cabo sin que aparezca necrosis. Estudios en animales anestesiados con el tórax abierto, sometidos de una a cinco horas de oclusión coronaria parcial han demostrado que el corazón verdaderamente se adapta a una reducción sostenida del flujo miocárdico en reposo (26). Muchos investigadores han reportado el desarrollo exitoso de un acoplamiento sostenido entre una baja perfusión y la contracción (también llamada hibernación a corto plazo), tanto en corazones de perro como de cerdos. La isquemia se produjo por una oclusión coronaria incompleta que llevó a una reducción del flujo miocárdico transmural del 20 % al 70 %. A pesar de una continua disminución en el flujo y al mismo tiempo de una continua disfunción sistólica, se observó un fenómeno intrigante, el cual consistió en la aparición de una resolución espontánea de algunos de los marcadores metabólicos de isquemia, principalmente a una disminución de la producción de lactato y la regeneración del fosfato de creatina a niveles cercanos a la normalidad. La hibernación a corto plazo es una condición frágil e inestable, que si es sometida a la superimposición de una carga cronotrópica o inotrópica, resulta invariablemente en un incremento de la producción de lactato, disminución del fosfato de creatina y eventualmente necrosis miocárdica (27). No obstante, del hallazgo mencionado es posible mantener un equilibrio precario entre un aporte disminuido de oxígeno y una disminución en la demanda del mismo para sustentar por algún tiempo la situación precaria mencionada, que puede ser de semanas o meses. Se trata de un acoplamiento entre perfusión y función contráctil que constituye el tejido atontado y consecuentemente, de persistir las mismas alteraciones metabólicas, el tejido hibernado.
Nuestro estudio se basa en los resultados del tratamiento médico exclusivamente, lo cual nos ha facilitado la observación clínica y electrocardiográfica de diferentes estados de atontamiento e hibernación resueltos favorablemente, sin tener que llegar a estudios muy especializados, como son eco sensibilizado con atropina y dobutamina, estudios nucleares y tomografía con emisión de positrones, hemodinamia, ultrasonido intracoronario y otros, tal como ha sido planteado por otros autores para descubrir su presencia. Sin embargo, las primeras observaciones, que fueron la base de este conocimiento, tropezaron con muchas dificultades, como el efecto del volumen parcial, la disminución del grosor (20 % a 25 %) del tejido hibernado, razón por la cual se sobrestima el flujo regional, la contradicción entre la moderada alteración regional residual y los hallazgos estructurales, y por último, la presencia previa, en los pacientes estudiados, de infarto del miocardio. Vanoverschelde (24) observó en 26 pacientes con enfermedad arterial coronaria sin infarto del miocardio previo, pero con oclusión completa de una arteria coronaria mayor comprobada por angiografía coronaria, que estos pacientes no tuvieron zona inactivable por haber estado sometidos a lo que hoy se conoce como precondicionamiento (períodos cortos de isquemia favorecen la posibilidad de soportar períodos largos de isquemia, debido a la estimulación de adenosina en los períodos cortos y a la consecutiva apertura de arteriolas coronarias menores de 200 micras de diámetro), que produjo circulación colateral suficiente. Estos pacientes fueron subdivididos en dos grupos, uno que correspondió a los que tenían motilidad en reposo normal, y otro a los que la presentaron anormal. Los que tuvieron motilidad en reposo normal no presentaron diferencia significativa en el flujo miocárdico entre la pared normal y el miocardio dependiente de circulación colateral. Por otra parte, los pacientes con anormalidad en la motilidad en reposo, el flujo miocárdico fue mayor en la pared normal que en la porción de la pared que tenía circulación colateral. Lo sorprendente de este estudio es que no se encontró diferencia significativa en el flujo entre los segmentos dependientes de colaterales de ambos grupos.
Para corroborar estos resultados se ha investigado también el consumo de oxígeno utilizando tomo-grafía por emisión de positrones. En los pacientes con motilidad normal de la pared en reposo, el consumo de oxígeno fue comparable entre el tejido normal y el tejido con circulación colateral. En los pacientes con anormalidad de la pared en reposo, el consumo de oxígeno fue mayor en el miocardio normal que en el colateralizado. Sin embargo, en los tejidos colateralizados de ambos grupos, el consumo de oxígeno fue igual. ¿Entonces, si el flujo y el consumo de oxígeno no se reducen, qué se debe alterar para que ocurra una reducción crónica de la función mecánica, observada en los segmentos con circulación colateral y alteración en la motilidad? Vanoverschelde (24) utilizando dipiridamol, demostró que el aumento de flujo se produjo en el miocardio normal, y marcadamente menor en el colateralizado, razón por lo cual no hubo respuesta inmediata de la motilidad en este último segmento, que ellos explican por la presencia de tejido hibernado. De acuerdo a ello los autores sugieren que episodios repetidos y cortos de isquemia, seguido por atontamiento miocárdico, pueden ser un mecanismo que conduzca a una disfunción isquémica regional crónica (hibernación). Así como la disminución del flujo disminuye la función contráctil, ésta a su vez provoca disminución del flujo. 4 a 6 horas después de la reperfusión aún el flujo permanecía disminuido 20 % a 25 % en el miocardio atontado en forma aguda, mientras que la función contráctil se recuperó lentamente (25-27). En la isquemia inducida por aumento de la demanda de manera sostenida en cochinos, a pesar de no haber cambios en el flujo miocárdico durante el estrés, el flujo sanguíneo disminuyó después de cesar éste, y estuvo disminuido durante un período prolongado. Es posible que ello esté relacionado con alguna forma de atontamiento miocárdico, y se pueda sugerir que la disminución progresiva del flujo miocárdico bajo estas condiciones es de alguna manera secundaria a la reducción de la función contráctil en reposo.
Atontamiento o aturdimiento del miocardio (28-32)
En el tejido atontado no se observan cambios estructurales por ser más una alteración funcional, que electrocardiográficamente tienen su expresión máxima en el interior de la zona inactivable, mezcla de células fibrosadas o muertas e hibernadas (onda Q o equivalentes), tejido isquémico (alteraciones del ST y de la T), bloqueo de rama izquierda, alteraciones del ritmo, sobrecarga sistólica del VI en el hipertenso (grado III y IV de nuestra clasificación) (13), etc.
La disminución o suspensión del flujo sanguíneo miocárdico ocasionado por una obstrucción aterotrombótica genera los siguientes cambios en el área miocárdica correspondiente a la arteria obstruida: 1. A los 8 segundos el metabolismo aeróbico normal cambia al metabolismo glucolítico anaeróbico, con disminución o cesación de la contractilidad. Disminuye el ATP celular, se acumula ADP, disminuye la fosfocreatina (a los 30 segundos sólo queda el 10 % de ella) y pueden aparecer los primeros cambios en el ECG-12D (lesión e isquemia subendocárdica). 2. Un poco más tarde sucede la fase reversible (espontánea, por efecto trombolítico de plasminógeno tisular predominante sobre el PAI-1) de la isquemia, sin embargo, para entonces sólo queda el 20 %-25 % del ATP previo. La glicólisis anaeróbica sólo es capaz de proporcionar el 80 % de los fosfatos de alta energía (FAE), utilizando la glucosa-1-fosfato como sustrato, generando 3 µmol de FAE y 2 de lactato. Éste, sumado a los iones H+ se acumula y disminuye el pH (5,8), favoreciendo la sobrecarga de partículas osmóticamente activas para ocasionar un grado leve de edema intracelular, conjunto que constituye la causa del dolor precordial. Los H+ se intercambian con el Na+, el cual aumenta en el interior celular, al igual que el Ca++. A los 15 minutos de isquemia y 3-20 de reperfusión no se observa este aumento del calcio. Si se restablece la perfusión y la isquemia ha sido breve, se repone la normalidad, lo cual no quiere decir que se abandone el paciente, ya que se debe investigar la causa de lo sucedido y su tratamiento o prevención. Pero en ocasiones aun cuando ello sucede, no se ha logrado la completa recuperación funcional batmotropa, cronotropa e inotropa cardíaca, es decir, el corazón ha quedado aturdido, normalidad que puede tardar horas a semanas en lograrse. En otras palabras, ha quedado una discordancia entre perfusión y función. Según Bolli (27) hay disfunción pos-isquémica, que no es causada por una falla primaria de la perfusión, lo cual en nuestra opinión, se debe a una alteración metabólica, que sin embargo sí es consecuencia de esa falla primaria de la perfusión.
Por tanto, en la definición de tejido atontado o aturdido, debe presentarse bajo estas dos situaciones: 1. Ausencia de daño miocárdico irreversible (28). 2. Flujo coronario restablecido a corto plazo después del ataque que ocasionó la alteración funcional. En la producción del miocardio atontado se han propuesto varias hipótesis:
El calcio (27). Se presentan diferentes situaciones: a) La disminución de la respuesta de los miofilamentos al calcio ocasiona menor fuerza de actividad por este ion, así como también disminución de la sensibilidad del miocardio al calcio extracelular. Se opone a esta posibilidad el hecho que el miocardio aturdido responde normalmente o casi normalmente a los inotrópicos y porque el miocardio aturdido responde normalmente al calcio intracelular, lo cual significa que su sensibilidad al electrolito es normal. b) Sobrecarga del calcio. Durante la isquemia, probablemente por la disminución del ATP y en consecuencia de la bomba ATPasa de potasio/sodio-calcio ocurre un aumento transitorio del calcio intracelular, el cual vuelve a lo normal a los pocos minutos de la reperfusión, debido a la menor captación del calcio por el retículo sarcoplásmico. Sin embargo, de mantenerse la isquemia, puede ocurrir la inundación del calcio intracelular y la muerte de ella. De esta manera, al quedarse el calcio en el citosol pueden activar proteinquinasas, fosfolipasas y enzimas degradativas que pudieran provocar una disfunción contráctil, igualmente falta de acoplamiento entre la excitabilidad y la contractilidad por disfunción del retículo sarcoplásmico. Los bloqueantes de los canales de calcio han demostrado aumento de la función contráctil en el miocardio aturdido de animales intactos (26). En cuanto al calcio, Atar y col. (27) distinguen la alteración de la contractilidad en el aturdimiento miocárdico pos-isquémico, cuya variación principal se debe a la injuria de las proteínas contráctiles por las proteasas durante la reperfusión (discordancia entre el calcio y el estado contráctil), de la observada en la insuficiencia cardíaca (IC), que se debe a un mal manejo del tránsito del calcio por disfunción del retículo sarcoplásmico, observándose además alteración de la expresión de genes y cambios de isoformas de miosina de los miofilamentos. Es decir, que en la IC hay cambios en la homeostasis del calcio intracelular y de la función de los filamentos. El aturdimiento pos-isquémico, sin embargo, no es tan fácil de explicar, ya que se atribuyó a una reducción de ATP, lo cual fue descartado porque dicha disfunción ocurre antes de la aparición de cambios del ATP y porque su disminución conduciría a la rigidez de la fibra, en vez de la pérdida de tensión de la pared muscular. Se han propuesto otras hipótesis: cambio de energía libre en la hidrólisis del ATP, disminución de la tasa de refosforilación del ADP citosólico, acidosis intracelular, acúmulo de fosfato inorgánico y transporte deficiente del calcio.
Los radicales libres de oxígeno (28-30). Los más estudiados han sido el superóxido, el hidroxilo y el precursor de RLO, el peróxido de hidrógeno. A los 15 minutos de isquemia ya hay RLO, pero su pico se produce durante la reperfusión (hasta las 6 horas), que es máxima a los 2-3 minutos de haberse iniciado. Se ha observado que a mayor cantidad de RLO mayor severidad de la disfunsión contráctil del miocardio. También que los bloqueantes de calcio aumentan la fuerza contráctil en el miocardio atontado. Los antioxidantes, los barredores de RLO y los protectores de sus efectos deletéreos como la trimetazidina mejoran la función contráctil del miocardio en el tejido atontado (13). El mecanismo de acción de los RLO es el siguiente (13): 1. Desnaturalizan las proteínas. 2. Inactivan las enzimas. 3. Peroxidan la membrana celular y dejan salir potasio, aun cuando la ATPasa de Na/K se haya recuperado. 4. Alteran la permeabilidad celular y la función de los organelos. 5. Atacan el sarcolema, ocasionando interferencia en el transporte de calcio. 6. Interfieren la ATPasa de calcio y de Na/K. 7. Alteran la función contráctil. 8. Alteran la función del retículo sarcoplásmico.
El papel del potasio (13,15). Su disminución en el interior celular es la causa principal de todos los trastornos, que puede ser general (hipokalemia sistémica) o regional cuando se instala la isquemia en una región y se desarrolla el mecanismo de producción íntimo del aturdimiento. Este mecanismo comprende: 1. Interferencia en la recuperación del potasio durante el potencial de reposo transmembrana (PRT) o fase 5. 2. Disminución del PRT y del potencial umbral (PU). 3. Pendiente mayor del PU. 4. Aparición de pospotenciales en las células de Purkinje auricular y ventricular. 5. Aparición de focos de mayor excitabilidad en el miocardio común auricular y ventricular (arritmias). 6) Disminución del potencial de acción transmembrana (PAT) en altura (disminuye el voltaje) y en tiempo con desaparición de la meseta, causa de disfunción contráctil por menor entrada de calcio al interior celular (paresia). De continuar la disfunción contráctil y los factores causales (atontamiento en período agudo y subagudo) se producen daños estructurales, constituyéndose, aparte de la necrosis y de la apoptosis, la hibernación. Entonces la paresia miocárdica pasa a hipoquinesia y ésta a aquinesia. Esta es la evolución de la angina, del bloqueo de rama izquierda, de los cambios de ST y T, de los bloqueos AV, de los trastornos del ritmo por formación, de las arritmias (trastornos de origen) y de otros trastornos eléctricos como consecuencia de la hipokalemia. En los casos agudos la evolución se hace en horas por alteración de mayor intensidad y extensión de los daños, llegándose a las alteraciones estructurales en pocas horas (infarto del miocardio). Debe distinguirse además, de acuerdo al tejido afectado así como también del daño estructural que continúa al miocardio atontado, es decir, el miocardio hibernado, la reacción esperada. Así, si el daño se encuentra en las células de Purkinje auricular o ventricular (tejido automatizado), provocará pospotenciales, que pueden ser el inicio de latidos prematuros si además encuentra, por el mismo mecanismo de la hipokalemia, pero ahora en el miocardio común (auricular o ventricular), un foco de excitabilidad aumentada con disminución del potencial umbral, lo cual proporciona el referido aumento de la excitabilidad y facilidad de respuesta del mismo (extrasístoles auriculares o ventriculares). Hasta este momento podría tratarse de atontamiento, pero si la isquemia y la hipokalemia regional aumentan podría llegarse a la alteración estructural que conduce al mecanismo de la reentrada y producirse trastornos de origen del ritmo cardíaco, es decir, arritmias supraventriculares y ventriculares del tipo de las taquicardias supraventriculares y ventriculares. Por lo general, para que se produzca este último mecanismo debe agregarse la infiltración colágena, lo cual es consecuencia de la hipertensión arterial por efecto de la aldosterona, cuando esta enfermedad ha sido la causa de la CIM aguda o subaguda.
Causas que contribuyen a la producción de miocardio aturdido y mantienen la isquemia miocárdica en fase aguda (funcional), lo cual repercute negativamente en el establecimiento de miocardio hibernado en fase crónica (estructural).
1. Intensidad y duración de la isquemia.
2. Intensidad y duración de los RLO en la membrana celular, especialmente durante la reperfusión rápida.
3. Temperatura del miocardio.
4. Área cardíaca afectada grande.
5. Carga del corazón: precarga (volumen) y poscarga (enfermedad hipertensiva esencial). Obesidad y otros.
6. Número de episodios de isquemia por día (3 o más). Agregación plaquetaria y vasoespasmo mantenidos.
7. Extensión y rapidez de la aterosclerosis. Factores de riesgo mayores persistentes.
8. Extensión y rapidez de la trombosis. Circulación colateral deficiente. Sedentarismo debido a falta de precondicionamiento (efecto que se pierde por el café). Diabetes. IM en jóvenes.
9. Frecuencia cardíaca mayor de 90 y menor de 40 por minuto.
10. Aumento de las catecolaminas (estrés, drogas, alcohol, nicotina, café).
11. Trombosis (PAI-1) > trombólisis (plasminógeno).
12. Hipokalemia e hipomagnesemia. Diuréticos perdedores de potasio y magnesio.
13. Acidosis metabólica (dolor precordial, aumenta hipokalemia intracelular).
14. Enfermedad hipertensiva esencial (EHE). Hipertrofia ventricular izquierda desde grado II / VIII, según clasificación de Bermúdez Arias (13,15).
15. Exceso de sodio, calcio y grasas saturadas.
16. Aumento de la viscosidad sanguínea. Hipoxia crónica. Vitamina B12.
17. Cambios ECG-12D recientes y mantenidos o evolutivos hacia el empeoramiento crónico.
18. Evolución natural sin modificación con tratamiento médico-dietético-farmacológico causal o con tratamiento médico convencional.
El miocardio hibernado (30-41)
Esta entidad ha sido más difícil de comprender. Corresponde a una disfunción miocárdica instalada desde el principio de la isquemia y su consecutivo cambio molecular, en una entidad que además es crónica en su evolución, pero reversible a cualquier edad después de su inicio mediante reperfusión y protección molecular. Se trata de miocitos comunes (los más frecuentemente involucrados) o automatizados que son viables pero no funcionantes (30).
La relación de intersecancia de las causas de la CIM demuestra que existe estrecha asociación entre el flujo coronario, el consumo miocárdico de oxígeno y la utilización metabólica como manifestación de balance adecuado con el desempeño contráctil del corazón, lo cual constituye el verdadero principio fundamental de la fisiología cardíaca total en esta área (17). Debido a la pequeña capacidad que tiene el miocardio de extraer oxígeno de sus reservas (mioglobina), una disminución del flujo sanguíneo coronario, se traduce rápidamente en una disminución del desempeño funcional del miocardio. Otras investigaciones han examinado en perros sometidos a disminución progresiva del flujo coronario la relación entre el flujo sanguíneo miocárdico regional y su función contráctil. Estos estudios han demostrado la existencia de una relación muy cercana, o mejor dicho, de un acoplamiento entre el suministro de substratos del miocardio (incluyendo oxígeno) y la demanda miocárdica de energía. La disminución proporcional entre el flujo miocárdico y su función contráctil ha sido llamada "acoplamiento agudo entre la perfusión y la contracción".
Por otra parte, la reperfusión después de períodos cortos de isquemia (menos de diez minutos), usualmente concluye en una rápida y completa restauración de las funciones cardíacas (32,33), con mantenimiento normal de la ultraestructura miocárdica (34). La hibernación miocárdica ocurre como resultado de una respuesta adaptativa a una reducción sostenida e importante del flujo miocárdico en reposo, tal como se observa en la isquemia miocárdica severa crónica, con infarto del miocardio previo o sin éste.
Bajo condiciones agudas de isquemia la contracción miocárdica se acopla de manera proporcional al suministro sanguíneo disponible para un momento dado (35,36). Esto ha inducido a los investigadores a examinar si un acoplamiento perfusión/contracción sostenido podría llevarse a cabo sin que aparezca necrosis. Estudios en animales anestesiados con el tórax abierto, sometidos de una a cinco horas de oclusión coronaria parcial han demostrado que el corazón verdaderamente se adapta a una reducción sostenida del flujo miocárdico en reposo (36). Muchos investigadores han reportado el desarrollo exitoso de un acoplamiento sostenido entre una baja perfusión y la contracción (también llamada hibernación a corto plazo), tanto en corazones de perro como de cerdos (28). La isquemia se produjo por una oclusión coronaria incompleta que llevó a una reducción del flujo miocárdico transmural del 20 % al 70 %. A pesar de una continua disminución en el flujo y al mismo tiempo de una continua disfunción, se observó un fenómeno intrigante, el cual consistió en la aparición de una resolución espontánea de algunos de los marcadores metabólicos de isquemia, principalmente la producción de lactato y la regeneración del fosfato de creatina a niveles cercanos a la normalidad. La hibernación a corto plazo es una condición frágil e inestable, que si es sometida a una superimposición de una carga cronotrópica o inotrópica, resulta invariablemente en un incremento de la producción de lactato, disminución del fosfato de creatina y eventualmente necrosis miocárdica (37-39). La hibernación a corto plazo (30,40) sucede cuando el acontecimiento isquémico (con poco flujo residual) es de aparición súbita, dura más de 20 minutos y sucede en la angina de pecho inestable y en el infarto del miocardio (35-38). La hibernación a largo plazo es el resultado de procesos isquémicos crónicamente mantenidos (39) con intensidad y tiempo variables, que van modificando la fisiología cardíaca permanente y paulatinamente (40), pero que de todos modos, se las ingenia para preservar algunos miocitos en condiciones de viabilidad, para lo cual es necesario la disminución de su potencial de acción y en consecuencias de todas sus funciones, y no sólo de la contráctil (41,42).
No obstante los hallazgos mencionados, es posible mantener un equilibrio precario entre un aporte disminuido de oxígeno y una disminución en la demanda del mismo para sustentar por algún tiempo, que puede ser de semanas, meses o años, el acoplamiento entre la perfusión y las funciones cardíacas, aunque ambas disminuidas. Ferrari (38) considera que la hibernación es un estado singular de isquemia miocárdica, en el cual los miocitos tienen su contractilidad disminuida pero permanecen viables, y en el que la revascularización obtiene la recuperación funcional, a diferencia de lo que ocurre cuando el tejido se ha necrosado. Contrariamente, los miocitos hibernados, en los casos de infartos del miocardio antiguos estudiados por nosotros, pueden quedar englobados en el interior de los miocitos fibrosados, es decir, dentro del área registrada por el ECG-12D como zona eléctricamente inactivable, lo cual significa, que en las fibras miocárdicas hibernadas no solamente disminuye su contractilidad, sino también la despolarización, que posiblemente sea el origen de todo el proceso. En efecto, en nuestros casos el orden de mejoría o recuperación de la función y estructura miocárdicas en cuanto al tiempo es el siguiente: 1. Desaparición del dolor precordial (97 %). 2. Mejoría o desaparición del desnivel del segmento ST (lesión subepicárdica en la mayoría de los casos, 92 %). 3. Disminución o desaparición de la negatividad de la onda T (isquemia subepicárdica en la mayoría de los casos, 92 %). 4. Disminución o desaparición de la onda Q patológica o sus equivalentes (33,4 %). Estos hallazgos se muestran en los electrocardiogramas representados en las figuras 2 a 7. Corresponden a electrocardiogramas de enfermedad hipertensiva esencial, y de cardiopatía isquémica metabólica, algunos con insuficiencia cardíaca, que han respondido favorablemente al tratamiento antihipertensivo y antiisquémico, por recuperación del miocardio hibernado. Cada ECG-12D tiene la interpretación correspondiente. De cada caso se analiza el electrocardiograma antes y después del tratamiento.
La diferencia con Ferrari probablemente se deba a que dicho autor se basó en la respuesta positiva a la dobutamina del tejido hibernado no necrosado, mientras que nosotros la hemos fundamentado en la respuesta directa al tratamiento revascularizante (con antiagregantes tipo heparina de bajo peso molecular o HBPM, indobufeno, clopidogrel) por la ruta metabólica glucolítica (y no por la de los ácidos grasos), la cual proporciona moléculas de ATP con el menor consumo de oxígeno (trimetazidina), e igualmente bajo protección miocárdica a los RLO, analizando la respuesta clínica y electrocardiográfica. Observaciones similares no prospectivas fueron reportadas posteriormente (1989 y 1997) por Rahimtoola (23), pero en ausencia de isquemia aguda y de necrosis miocítica. No se sabe con certeza su etiología ni su fisiopatología, pero se piensa que corresponde a un mecanismo de protección endógeno (43-51). Según Hearse (52) la posibilidad de su existencia es apenas del 50 %, mientras que para otros autores se encuentra entre el 25 %-40 %. En ellas existe disminución de la contractilidad, y en menor proporción la posibilidad de alteraciones de las otras funciones cardíacas, paralelamente a la disminución mantenida del flujo sanguíneo.
El atontamiento miocárdico agudo o subagudo con su disfunción diastólica y sistólica del ventrículo izquierdo (11,12), contribuye en una forma u otra a la disfunción contráctil crónica del miocardio hibernado (44-48). Esta alteración puede ser tan variable en su expresión clínica, electrocardiográfica y de otros métodos, que puede ir desde el tejido hibernado no evidenciable por las exploraciones actuales, incluyendo la tomografía de emisión de positrones (TEP) (37), hasta alteraciones tan grandes como la zona inactivable (38), la isquémica, la insuficiencia cardíaca, las alteraciones del ritmo, las cuales pueden ser fácilmente detectadas por los métodos habituales, especialmente por el ECG-12D. Y este es precisamente el propósito de este estudio: demostrar la evidencia de tales alteraciones y su reversibilidad mediante tratamiento médico farmacológico, con la objetividad representada por la mejoría clínica y electrocardiográfica, demostrada por este último método de manera inobjetable (13,15,19,20) y sostenida en la práctica clínica por más de 15 años, de 1984 a 1998 y publicada por primera vez en la literatura mundial en 2000 (13). Los resultados se mantienen en el mismo orden hasta el momento. Se podría decir, en conclusión, que el "corazón es astuto", porque logra alcanzar en la mayoría de los casos estudiados por nosotros un equilibrio entre perfusión y función, y que este equilibrio se mantiene a través del tiempo, esperando la ayuda del que haya comprendido este estado de "inteligencia natural" del corazón isquémico, ayuda que obviamente está en la reperfusión lenta bajo protección miocárdica, pasando de algún modo nuevamente por el aturdimiento, hasta llegar a la recuperación parcial o total, pero que, aunque parcial o total, de todos modos altamente favorable para el paciente, porque puede ser logrado mediante tratamiento médico dietético farmacológico causal, sin que tenga que concurrirse al intervencionismo ni a la cirugía, como hasta el presente se ha concebido, y que en vez de beneficiarlo, lo altera más.
En 1978 Diamond y col. (22) sugieren que el miocardio isquémico no infartado (en nuestros casos se incluyeron los casos infartados recientes y antiguos) puede vivir en un estado de hibernación funcional. Rahimtoola (23), en 1989 propuso el concepto del miocardio hibernado al plantear la posibilidad de revertir por revascularización a la insuficiencia cardíaca congestiva (ICC) de origen isquémico. Condición que según el autor se debía a una respuesta poco común, o sea, a la reducción de su función hasta lograr el equilibrio de ésta con el flujo miocárdico disminuido en reposo, sin que se presente (o aumente si ya existe) necrosis ni nuevos síntomas (que ya podían estar presentes). En paréntesis nuestras observaciones. Ahora bien, para que ese mecanismo protector suceda espontáneamente, no solamente debe suceder los cambios funcionales descritos, sino también cambios estructurales y bioquímicos endógenos (49-52), como se describirán más adelante.
Para Vanoverschelde y col. (24) el concepto de hibernación crónica tiene importancia para el manejo de la CIM crónica, al establecer que la disfunción contráctil se debe a la reducción mantenida del flujo coronario. Opinamos que es importante en todas las fases (aguda, subaguda y crónica) de la CIM, por lo cual preservar la estructura y la función del miocardio en cualquiera de esas fases evolutivas, adquiere la mayor importancia, debido a que a mayor protección y reperfusión, más posibilidad que las fibras hibernadas superen a las fibrosadas, aún en casos de infarto del miocardio y de ICC.
¿Cómo pueden mantenerse los miocitos hibernados viables? Esta respuesta tiene varias opciones razonables: 1. Que no haya nuevas crisis isquémicas, por lo cual la dieta y el tratamiento médico debe mantenerse bajo dosis mínimas que garanticen la ausencia de esas crisis (16). 2. Que se establezca un cerco protector alrededor de la mayor concentración de ellas en el área isquémica o necrosada, lo cual sólo es factible mediante la apoptosis, por sufrir estas células la muerte no inflamatoria, como sucede en las que mueren por inundación de calcio debido a la isquemia. Este efecto, planteado por nosotros para explicar la conservación de los miocitos hibernados a través del tiempo (20), también lo exponen Lim y col. (54), que plantean la existencia de apoptosis en el corazón hibernado al admitir definitivamente la presencia de pérdida de miocitos sin fibrosis. 3. Hipertrofia compensadora de los miocitos. 4. Aumento mínimo o ausente de tejido conectivo miocárdico, por efecto de la aldosterona.
Cambios estructurales en la hibernación (33,54-56).
Se ha demostrado en el miocardio isquémico cambios estructurales del miocito y de la matriz celular en los segmentos más afectados crónicamente. Los hallazgos fueron los siguientes: pérdida del material contráctil, que a su vez pueden ser: a) En unas células en la región cercana al núcleo. b) En otras, en una porción más extensa, con destrucción de sarcómeros y muy pocos de ellos en la periferia celular. La pérdida miofibrilar no se acompaña de cambios mayores intracelularmente, lo cual es evidentemente diferente a la degeneración atrófica. c) El espacio que estaba ocupado por miofilamentos está ahora lleno de glucógeno. d) Las mitocondrias son pequeñas y ocupan todo el citoplasma miolítico. e) El núcleo es tortuoso y muestra heterocromatina. f) Por último el retículo sarcoplásmico y los túbulos están ausentes. No obstante, no hay signos de degeneración, tales como vacuolización citoplasmática, edema intracelular, disgregación mitocondrial o ruptura de la membrana, así como tampoco, presencia de vesículas citosolares lipídicas que pudieran indicar daño agudo isquémico o atrofia celular.
Alteraciones bioquímicas (34-36)
Se conocieron antes que los cambios estructurales. El contenido en adenosintrifosfato (ATP), adenina y fosfato de creatina está muy cercano a la normalidad. La relación adenosintrifosfato/adenosindifosfato (ATP/ADP) y fosfocreatina/adenosintrifosfato (PC/ATP), que reflejan la función mitocondrial, se encuentran casi normales e indican que el consumo de oxígeno está presente en condiciones de normalidad en el miocardio atontado y en el hibernado. En ayunas el tejido hibernado posee más glucosa que el normal. Estos hallazgos se han invocado para explicar la reversibilidad de la disfunción regional del tejido hibernado después de la revascularización. Comparando los hallazgos morfológicos con los metabólicos ha surgido la pregunta sobre la utilización de la glucosa por parte de las células hibernadas. Originalmente se pensó que la presencia de glucosa en este tejido reflejaría la ausencia de estimulación anaeróbica en la isquemia crónica, pero, ¿cómo aceptar esta respuesta sí el flujo sanguíneo miocárdico y el consumo de oxígeno están cercanos a lo normal? ¿Cómo postular el incremento de glucógeno cuando se esperaría lo contrario? La activación crónica de la glucógenosintetasa por la isquemia se ha implicado como uno de los cambios que dan cuenta de las alteraciones metabólicas observadas en la hibernación miocárdica.
Alteraciones moleculares (34-42,57)
En efecto, McNulty y Luba (42) han demostrado que la isquemia transitoria induce a una activación sostenida de la glucógenosintetasa independiente-mente de la glucosa 6-P, lo que ayuda a una rápida recuperación de los depósitos de glucógeno durante la reperfusión. De esta manera todos los hallazgos apoyan la idea que las alteraciones en el metabolismo de la glucosa vistos en la isquemia experimental y en la hibernación puede ser el resultado de una disregulación sincronizada de la glicólisis y de la glucogenólisis. Finalmente, como en estos casos se observa un aumento en la captación de la glucosa, independientemente del entorno hormonal, algunos autores han postulado que el aumento de la incorporación de esta sustancia puede deberse a un cambio en la expresión de los Gluts 1 y 4 (transportadores transmembrana de glucosa 1-4) que predominan en el corazón normal a Gluts 3 y 2, (transportadores transmembrana de glucosa 3-2) que son relativamente independientes a la presencia de la insulina. Estudios recientes han sugerido que los cambios estructurales que se observan en el miocardio hibernado son consecuencia de una serie de procesos de desdiferenciación. En efecto, los miocardiocitos hibernados muestran muchas de las características moleculares de los miocardiocitos neonatales, entre ellos: 1. Depleción de los filamentos contráctiles. 2. Presencia de retículo sarcoplásmico rugoso. 3. Acumulación de glucógeno. 4. Núcleo de forma irregular. 5. Pérdida de la organización del retículo sarcoplásmico. 6. Pérdida de los túbulos T y vesiculización del sarcolema. No todas estas alteraciones recuerdan a las células fetales. Así por ejemplo: 1. Los sarcómeros están sin ordenación absoluta en la periferia celular del tejido hibernado, mientras que en el tejido fetal lo están de manera aleatoria. 2. La cantidad de glucógeno es mayor que en miocardio fetal. 3. La hipótesis de la diferenciación también está apoyada por estudios inmunohistoquímicos que muestran que los miocardiocitos hibernados reexpresan proteínas contráctiles que son específicas del corazón fetal, como son la L-Actina, propia del músculo liso fetal, presente en el miocardio en esa etapa de la vida, mientras que al mismo tiempo exhibe la misma organización de ciertas proteínas estructurales, como la lentina, tal como ocurre en los miocardiocitos inmaduros (40-46). 4. Aparecen otras nuevas proteínas, como la cardiotina, una proteína de alto peso molecular ausente en las células fetales y también en las hibernadas. Todo indica que las células hibernadas están sometidas a una desdiferenciación parcial (45). 5. La oclusión parcial coronaria durante 15 minutos y reperfusión a través de una estenosis crítica, da lugar a disminuciones concordantes de flujo y función (hibernación) después de una semana, dejando de lado aquellas críticas sobre falta de estudios experimentales de hibernación crónica. También encontraron signos de miolisis y aumento de contenido de glucógeno en el miocardio (40). 6. El miocardio disfuncionante tiene disminución de las proteínas SERCA2a (transportadora de calcio en el retículo endoplásmico 2a) y fosfolamban, lo cual ha dado lugar a la especulación de la existencia de alteración éxcitocontráctil, hallazgo considerado por algunos como casi patognomónico de miocardio hibernado (52).
Los eventos moleculares que desembocan en el fenotipo hibernado aún no han sido totalmente esclarecidos, debido a que la isquemia y los RLO juegan un papel crucial en el cambio del miocito normal a miocito hibernado, pasando previamente por los cambios funcionales del miocito atontado. El tejido hibernado forma parte estructural de la lesión, de la isquemia, de la zona inactivable del miocito contráctil, y también de los daños estructurales del sistema automatizado, como en el bloqueo de rama izquierda con eje a la izquierda de +60º (descrito por Dhingra en 1978 (31) y derecha, enfermedad del nodo sinusal, trastornos de la conducción en el nodo AV y otros menos frecuentes, todos ellos consecutivos a alteraciones del potencial de acción transmembrana, que se hace menos intensa (menor voltaje) y más angosta (menor contractilidad) en los miocitos contráctiles y en los automatizados. El bloqueo de rama izquierdo el eje de QRS a la derecha de +60º empeora el pronóstico, según observaciones de Francisco Arocha Valbuena: pronóstico del paciente con bloqueo de rama izquierda con eje del QRS desviado a la derecha de más 60º (Tesis doctoral, Universidad del Zulia, 1998).
Las células hibernadas comparten con el tejido fibrosado la zona inactivable (onda Q patológica del ECG-12D, y con otras alteraciones menores el resto de los acontecimientos isquémicos demostrables mediante este método. Estos hallazgos han proporcionado la tesis que al ser el tejido hibernado parecido o similar al tejido miocárdico fetal del ventrículo izquierdo, que como se sabe es viable pero no funcionante por no participar en la circulación fetal, representa cambios estructurales (42-45) profundos de una verdadera protección endógena, presente además en todo tejido miocárdico isquémico que haya podido sobreponer la viabilidad funcional a la muerte.
Por tanto carece de sentido intentarlo por vía intervencionista, que en sí misma representa mayor daño endotelial, y también por el camino de la cirugía, a menos que sea la única alternativa después de haberlo intentado por lo menos durante tres meses, bajo la garantía de haber observado la ausencia casi absoluta de la muerte súbita una vez instalado el tratamiento dietético farmacológico causal, lo cual da oportunidad, de no mejorar y mantenerse el paciente en condiciones de control asintomático, a programar la cirugía, cuyos mejores resultados lo proporciona la implantación de la mamaria a la descendente anterior pos-diagonal, preferiblemente sin circulación extracorpórea.
Por lo expuesto, carece de importancia el enfoque de diagnosticar la presencia del tejido hibernado, ya que él se encuentra allí, en el corazón isquémico que ha sobrevivido. Por ello estamos de acuerdo con Amstrong (citado por Camici (54), quien dice que el estándar apropiado para determinar viabilidad no es la presencia de actividad metabólica por TEP ni la perfusión por redistribución de talio o el incremento sistólico durante el ecocardiograma estrés con dobutamina, el estándar adecuado es la recuperación funcional y reducción de síntomas después de la revascularización. A lo cual agregamos: y la recuperación de la lesión, de la isquemia y de la zona eléctricamente inactivable (ZEI) electrocardiográfica, por ser ésta una manifestación, aunada a la recuperación clínica, definitivamente objetiva de la recuperación del miocardio, que a todas luces sólo podría ser por la mejoría del miocardio hibernado.
Algunos autores se preguntan si el flujo sanguíneo se encuentra disminuido o conservado, cuestión que en sí misma depende tanto de la arteria originalmente obstruida parcialmente (perfusión disminuida) u obstruida totalmente con IM, entonces la posible disminución de la perfusión dependería del resto de las arterias coronarias. En ambos casos la probabilidad de miocardio hibernado es casi del 100 %, como fue demostrado en nuestros casos, en los cuales la mejoría clínica, por recuperación del tejido hibernado mediante la reperfusión medicamentosa aconteció y se mantuvo durante 12 años en el 97 % de ellos. Igualmente influye, tanto en la presencia de miocardio aturdido como hibernado, la presencia de nuevos episodios isquémicos. La concordancia entre el flujo y la función cardíaca disminuidos, indica la posibilidad de la existencia de miocardio hibernado, sin embargo, su ausencia, reflejada por la gravedad del caso, no necesariamente la excluyen. Basados en la primera posibilidad, numerosos autores han intentado descubrir el tejido hibernado antes de intentar la reperfusión intervencionista (la mayoría de ellos) o medicamentosa, como es nuestra propuesta y experiencia.
Camici y col. (54) han señalado las siguientes características para la distinción del miocardio hibernado: 1. El flujo miocárdico basal está crónicamente disminuido en magnitud suficiente como para deprimir la función miocárdica, siendo ésta una condición sine qua non para el diagnóstico. 2. Hay consecuencias de la isquemia tisular, tal como remodelado sin necrosis. 3. Puede demostrarse reserva contráctil en segmentos hibernados. 4. Recuperación de la función deprimida luego de la revascularización, que en nuestra experiencia es la más importante y demostrativa, porque en sí misma contiene la solución, por ser tanto diagnóstica como terapéutica. Destaca Camici que no hay modelos animales experimentales que permitan simular la condición.
Belziti (55) señala que las pruebas diagnósticas para la determinación de la viabilidad miocárdica son: 1. Evocadoras de isquemia: Prueba de esfuerzo. Eco-estrés. Ejercicio. Dipiridamol. Dobutamina. Arbutamina. Radioisótopos: Perfusión miocárdica. Ventriculograma. 2. Optimizando las cargas ventriculares: Nitroglicerina (NTG). Potenciación extrasistólica. 3. Con inotrópicos: Dobutamina. Arbutamina. 4. Integridad vascular. Cinecoronariografía. Perfusión miocárdica radioisotópica: Talio 201, tomografía de emisión de positrones con acelerador lineal Sincrotrón (SPET), tecnecio (Tc) 99m, tomografía de emisión de positrones con acelerador lineal MIBI (MIBI SPET). 5. Actividad metabólica celular: Tomografía con emisión de positrones (TEP).
De cualquier manera se mantiene la controversia entre los que sostienen el concepto de hibernación como fenómeno independiente (23), al cual no unimos según lo demuestran los cambios estructurales, bioquímicos y moleculares, además de la respuesta recuperativa con el tratamiento médico propuesto en este trabajo y demostrado clínica y electrocardiográficamente. El otro grupo afirma que es simplemente consecuencia de episodios repetidos de aturdimiento miocárdico. Este grupo no reconoce la presencia de miocardio hibernado en casos de infarto del miocardio (56,57).
El precondicionamiento cardíaco (33,34,45-48) es una respuesta fisiológica recientemente descrita en el cual el compromiso isquémico del miocardio regula discretamente sus requerimientos energéticos y al mismo tiempo vuelve más tolerante al tejido contra la hipoxia más prolongada. En otras palabras, "períodos cortos de isquemia dan protección al miocardio para períodos más largos de isquemia". Se debe a que la adenosina, limita el influjo de calcio al interior celular, debido a una inducción primaria ocasionada por períodos cortos e intermitentes de isquemia seguida de reperfusión por vasodilatación espontánea ocasionada por la mencionada sustancia. ¿Por qué el precondicionamiento condiciona una contribución importante en la aparición de la hibernación miocárdica? 1. Hay evidencia que cambios en la expresión genética del miocardio, en especial el aumento de la síntesis de factores de crecimiento como el factor de crecimiento parecido a la insulina (IGF 1 y 2) y de factores de transcripción. Aunque estos argumentos son algo especulativos, ellos constituyen la base, al menos en parte, de la relación entre el precondicionamiento y la hibernación. 2) Otros factores no relacionados directamente con la isquemia, pero que sin embargo alteran crónicamente la fisiología cardíaca y por lo mismo contribuyen a la aparición del tejido hibernado son las arritmias (fibrilación auricular), el aumento de la demanda del ventrículo izquierdo por hipertensión arterial y la hipokalemia intracelular. La recuperación de estas alteraciones fisiopatológicas contribuyen a la recuperación y al mantenimiento del miocardio hibernado, especialmente cuando se logra el mecanismo del precondicionamiento, debido al desarrollo de la circulación vicariante o colateral bajo el poder vasodilatador de la adenosina generada en los períodos cortos de isquemia, los cuales se obtienen mediante caminatas de por lo menos 1 hora, en la tarde o noche, para evitar la susceptibilidad cardíaca del ritmo circadiano.
REFERENCIAS
1. Stamler J, Wentworth D, Neaton JD. Is relationship between serum cholesterol and risk of premature death from coronary heart disease continuous and graded? Findings in 356,222 primary screenees of the Multiple Risk Factor Intervention Trial (MRFIT). JAMA. 1986;256(20):2823-2838. [ Links ]
2. Anuarios de Mortalidad. Ministerio de Salud y Desarrollo Social. Caracas, 1998. [ Links ]
3. Larrea Sánchez JL. Tratamiento antitrombótico en el enfermo cardiovascular. Madrid: Editorial Doyma; 2000. [ Links ]
4. Kannel WB, McGee DL. Diabetes and glucose tole-rance as risk factors for cardiovascular disease: The Framingham study. Diabetes Care. 1979;2:120-165. [ Links ]
5. Remuzzi G. Endothelins in the control of cardiovascular and renal function. Lancet. 1993;342:589-592. [ Links ]
6. Jiang C, Sarrel PM, Poole-Wilson PA, Collins P. Acute effects of 17B-Estradiol on rabbit coronary artery contractile responses to endothelin-1. Am J Physiol. 1992;263:H271-H275. [ Links ]
7. Clavel A. Physiological significance of endothelium. Circulation. 1993;593:45-50. [ Links ]
8. Davies M. The vascular endothelium. Ann Surg. 1993;218(5):593-609. [ Links ]
9. Feldstein CA. Farmacología del endotelio. En: Soltero I, Cardona R, editores. Aterosclerosis al día II. Caracas: Editorial Greco; 1993.p.182-189. [ Links ]
10. Maseri A. The changing faces of angina pectoris: Practical implications. Lancet. 1983;1(8327): 746-749. [ Links ]
11. Braunwald E, Rutherford JD. Reversible ischemic left ventricular dysfunction: Evidence for the "hibernating myocardium". J Am Coll Cardiol. 1986;8:1467-1475. [ Links ]
12. Braunwald E, Kloner RA. The stunned myocardium: Prolonged, post-ischemic ventricular dysfunction. Circulation. 1982;66:1146-1149. [ Links ]
13. Bermúdez Arias F, Bermúdez Pirela V, Cano Ponce C. Cardiopatía isquémica. Caracas: Editorial McGraw-Hill Interamericana de Venezuela; 2000. [ Links ]
14. Bermúdez Arias F, Bermúdez Pirela V, Cano Ponce C. Modificación oxidativa de las lipoproteínas y su efecto aterogénico. En: Cardona R, Soltero I, editores. Aterosclerosis al día III. Caracas: Impresos Vigoni; 1996.p.163-171. [ Links ]
15. Bermúdez Arias F. Electrocardiografía diagnóstica. Caracas: Editorial McGraw-Hill Interamericana de Venezuela; 1998. [ Links ]
16. Bermúdez Arias F. Importancia de la dieta en la aterosclerosis y enfermedades cardiovasculares. En: Soltero I, editor. Aterosclerosis al día V. Caracas: Editorial Asociación Venezolana de Aterosclerosis-AVA; 2003.p.355-372. [ Links ]
17. Bermúdez Arias F. Angina de pecho e infarto del miocardio. México: Editorial Méndez Oteo; 1981. [ Links ]
18. Bermúdez Arias F, Velasco M. Indobufeno. Estudio de sus efectos clínicos y electrocardiográficos en 23 pacientes. Arch Ven Farmacol Terap. 1990;9:56-59. [ Links ]
19. Bermúdez Arias F, Baptista Arrieta G, Fernández A, Madrid L, Colmenares R, García F, et al. First American study of trimetazidine (Fast). Arch Ven Farmacol y Terap. 1996;14:345-356. [ Links ]
20. Bermúdez Arias F, Valmore-Bermúdez P, Clímaco-Cano P. Recuperación del miocardio hibernado. En Soltero I, editor. Aterosclerosis al día V. Caracas: Editorial Asociación Venezolana de Aterosclerosis-AVA; 2003.p.391-410. [ Links ]
21. Roan P, Scales F, Saffer S, Buja LM, Willerson JT. Functional characterization of left ventricular segmental responses during the initial 24 hours and one week after experimental myocardial infarction. J Clin Invest. 1979;64:1074-1088. [ Links ]
22. Diamond GA, Forrester JS, de Luz PL, Wyatt HL, Swan HJC. Post-extrasystolic potentiation of ischemic myocardium by atrial stimulation. Am Heart J. 1978;95:204-209. [ Links ]
23. Rahimtoola SH. The hibernating myocardium. Am Heart J. 1989;117:211-221. [ Links ]
24. Vanoverschelde JL, Wijns W, Borgers M, Heyndrickx G, Depre C, Flameng W, et al. Chronic myocardial hibernation in humans. From bedside to bench. Circulation. 1997;195:1961-1971. [ Links ]
25. Osakada G, Hess OM, Gallagher KP, Kemper WS, Ross J Jr. End-systolic dimension-wall thickness relations during myocardial ischemia in conscious dogs: A new approach for defining regional function. Am J Cardiol. 1983;51:1750-1758. [ Links ]
26. Bolli R. Mechanism of myocardial stunning. Circulation. 1990;82:724-734. [ Links ]
27. Atar D, Gao WD, Marban E. Alterations of excitation-contraction coupling in stunned myocardium and in falling myocardium. J Mol Cell Cardiol. 1995;27:783-791. [ Links ]
28. Chambers DE, Park DA, Patterson G. Xantino oxidasa as a source of free radical damage in myocardic ischemic. J Mol Cell Card. 1985;17:145-152. [ Links ]
29. López-Jaramillo P. Bioquímica del endotelio vascular: Implicaciones fisiológicas y clínicas. Función endotelial y radicales libres. Bucaramanga: Horizonte Impresores; 2001. [ Links ]
30. Ross J. Myocardial perfusion-contraction matching. Implications for coronary heart disease and hibernation. Circulation. 1991;83(3):1076-1083. [ Links ]
31. Dhingra RC, Amat Y, Leon F, Wyndham C, Sridhar SS, Wu D, et al. Significance of left axis deviation in patients with chronic left bundle branch block. Am J Cardiol. 1978;42:4551-4556. [ Links ]
32. Mills JT, Fallon D, Wrenn H, Sasken W, Gray J, Bier D, et al. Adaptive responses of coronary circulation and myocardium to chronic reduction in perfusion pressure and flow. Am J Physiol Heart. 1994;266:H447–H457. [ Links ]
33. Maes A, Flameng W, Borgers M, Nuyts J, Ausma J, Bormans G, et al. Regional myocardial blood flow, glucose utilization and contractile function before and after revascularization and ultrastructural findings in patients with chronic coronary artery disease. Eur J Nucl Med. 1995;22(11):1299-1305. [ Links ]
34. Cano C, Malik KU. Adenosine receptor-mediated alterations in prostacyclin production and cardiac function in the isolated rabbit heart. J Phar Exp Therap. 1992;261:660-668. [ Links ]
35. Hänsen P. Role of neutrophils in myocardial ischemia and reperfusion. Circulation. 1995;91:1872-1879. [ Links ]
36. Rinaldi C, Hall R. Myocardial stunning and hibernation. Indian Heart J. 1996;48(6):721-725. [ Links ]
37. Conversano A, Walsh JF, Geltman EM, Perez JE, Bergmann SR, Gropler RJ. Delineation of myocardial stunning and hibernation by positron emission tomo-graphy in advanced coronary artery disease. Am Heart J. 1996;131(3):440-450. [ Links ]
38. Ferrari R, Ceconi C, Curello S, Benigno M, La Canna G, Visioli O. Left ventricular dysfunction due to the new ischemic outcomes: Stunning and hibernation. J Cardiovasc Pharmacol. 1996;28(Suppl 1):18-26. [ Links ]
39. Heush G, Schulz R. Hibernating myocardium: A review. J Mol Cell Cardiol. 1996;28:2359-2372. [ Links ]
40. Schulz R, Guth BD, Pieper K, Martin C, Heusch G. Recruitment of an inotropic reserve in moderately ischemic myocardium at the expense of metabolic reco-very. A model of short-term hibernation. Circ Res. 1992;70(6):1282-1295. [ Links ]
41. Elsasser A, Schlepper M, Schaper J. Clinical and morphologic findings in the "hibernating myocardium" Zeistchchrift Kardiol. 1995;84(Suppl 4):107-114. [ Links ]
42. McNulty PH, Luba MC. Transient ischemia induces regional myocardial glycogen sintetasa activation and glycogen synthesis. Am J Phisiol. 1995;268:H364-H370. [ Links ]
43. Schwarz E. Myocite degeneration and cell death in hibernating human myocardium. J Am Coll Cardiol. 1996;27:1577-1585. (Abstract). [ Links ]
44. Chen Ma L, Linfert DR, Lai T, Fallon JT, Gillam LD, Waters DD, et al. Myocardial cell death and apoptosis in hibernating myocardium. J Am Coll Cardiol. 1997;30(5):1407-1412. [ Links ]
45. Ely S, Berne R. Protective effects of adenosine in myocardial ischemia. Circulation. 1992;85:893-900. [ Links ]
46. Cave AC, Silverman AS, Apstein CS. Ischemic preconditioning does not protect against contractile dysfunction in the presence of residual flow: Studies in the isolated, blood-perfused rat heart. Circulation. 1997;96(9):3087-3093. [ Links ]
47. Schulz R, Heusch G. Hibernating myocardium: No involvement of endogenous adenosine. Zeistchchrift Kardiol. 1996;85(Suppl 6):177-184. [ Links ]
48. Murry CE, Jennings RB. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124-1136. [ Links ]
49. James A, Fallavollita JA. Spatial heterogeneity in fasting and insulin-stimulated F-2-deoxyglucose uptake in pigs with hibernating myocardium. Circulation. 2000 (102): 908-914. [ Links ]
50. Rahimtoola SH. Importance of diagnosing hibernating myocardium: How and in whom? J Am Coll Cardiol. 1997;30:1701-1710. [ Links ]
51. Ferrari R. The search for the hibernating myocardium - Have we reached the limit? Cardiovascular Drugs and Therapy. 1999;13(2):137-143. [ Links ]
52. Hearse DJ. Myocardial hibernation. A form of endoge-nous protection? Eur Heart J. 1997;18(Suppl A):A2-A7. [ Links ]
53. Lim H, Fallavollita JA, Hard R, Kerr CW, Canty JM. Profound apoptosis-mediated regional myocite loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation. 1999;100:2380-2386.
54. Camici P, Rimoldi OE. Myocardial blood flow in patients with hibernating myocardium. Cardiovasc Res. 2003;57:302-311. [ Links ]
55. Belziti CA. Diagnóstico de viabilidad miocárdica. En: Oliveri R, editor. Viabilidad miocárdica. Buenos Aires: Ed. Médica Panamericana SA; 1999.p.211-226. [ Links ]
56. Boden WE, ORourke RA, Crawford MH. Outcomes in patients with acute non-Q-wave myocardial infarction randomly assigned to an invasive as compared with a conservative management strategy: Veterans affairs non-Q-wave infarction strategies in hospital (VANQWISH) trial investigators. N Engl J Med. 1998;338:1785-1792. [ Links ]
57. Anderson HV, Cannon CP, Stone PH, Williams DO, McCabe CH, Knatterud GL. One-year results of the thrombolysis in myocardial infarction (TIMI) IIIB clinical trial: A randomized comparison of tissue-type plasminogen activator versus placebo and early invasive versus early conservative strategies in unstable angina and non-Q wave myocardial infarction. J Am Coll Cardiol. 1995;26:1643-1650. [ Links ]
