











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkInvestigación Clínica
versión impresa ISSN 0535-5133
Invest. clín vol.55 no.3 Maracaibo set. 2014
Relevancia de la hiperglucemia para la evolución y el desenlace de la hemorragia subaracnoidea aneurismática: evidencias y mecanismos.
Iván Pérez-Neri.
Departamento de Neuroquímica, Instituto Nacional de Neurología y Neurocirugía. Ciudad de México 14269, México DF.
Autor de correspondencia: Iván Pérez-Neri. Departamento de Neuroquímica, Instituto Nacional de Neurología y Neurocirugía. Insurgentes Sur 3877. La Fama, Tlalpan Ciudad de México, México, D.F. Correo electrónico: ivanperezneri@hotmail.com
Resumen. La hemorragia subaracnoidea aneurismática (HSAa) puede tener un desenlace mortal en unas pocas semanas debido por las complicaciones que presenta, como el vasoespasmo y el edema cerebrales junto con la hiperglucemia. La hiperglucemia podría estar relacionada con el desarrollo del vasoespasmo y el edema cerebrales. Es posible que el control de la glucemia juegue un papel central en la evolución y el desenlace de la HSAa. Se han descrito los mecanismos por los cuales puede darse esta relación, que incluyen el equilibrio de iones, la liberación de aminoácidos excitadores, la estimulación de moléculas vasoconstrictoras y la disminución en la síntesis de vasorrelajantes. Sin embargo, existen estudios que no apoyan la hipótesis sobre la participación de la hiperglucemia en esta enfermedad. En conjunto, estas evidencias sugieren que el control de los niveles de glucosa podría modificar el desenlace de los grupos de pacientes con HSAa dependiendo de las complicaciones que presenten.
Palabras clave: isquemia, endotelina-1, óxido nítrico, metabolismo energético.
Relevance of hyperglycemia on the course and outcome of aneurysmal subarachnoid hemorrhage: evidence and mechanisms.
Abstract. Aneurysmal subarachnoid hemorrhage (aSAH) may have a fatal outcome after a few weeks from ictus, due to its complications, like cerebral vasospasm and edema along with hyperglycemia. Hyperglycemia may be involved in the development of brain vasospasm and edema. It is possible that hyperglycemia plays a central role in the outcome of aSAH. Several mechanisms may explain this relationship; they include ion balance, excitatory amino acid release, stimulation of vasoconstrictor molecules and reduced synthesis of vasorelaxants. However, some studies do not support this hypothesis regarding the role of hyperglycemia in aSAH. Taken together, the evidence suggests that the control of glucose levels may influence the aSAH outcome depending on the complications that may develop.
Keywords: ischemia, endothelin-1, nitric oxide, energy metabolism.
Recibido: 15-11-2013. Aceptado: 10-04-2014
INTRODUCCIÓN
La hemorragia subaracnoidea aneurismática (HSAa) representa el 5% de los eventos cerebrovasculares (1). Su incidencia es de 10 a 16 casos por 100.000 habitantes/año, incrementa con la edad (2) y ha aumentado a lo largo del tiempo mientras que su mortalidad ha disminuido a una tasa de 0,8% anual (3, 4). Es más frecuente en la circulación cerebral posterior y en las mujeres que en los varones y representa el 0,07% de todas las hospitalizaciones en los Estados Unidos (2-5). Se presenta comúnmente en la quinta década de la vida (4), pero puede ocurrir en sujetos más jóvenes.
Se caracteriza, al momento del ictus, por la aparición súbita de un dolor de cabeza intenso (1) y un conjunto de otros síntomas que puede variar según la localización del aneurisma (6); su desenlace puede ser muy favorable en algunos casos y muy desfavorable en otros (7) y puede resolverse en el curso de unas pocas semanas.
La estancia intrahospitalaria de los pacientes con HSAa puede durar entre 8 y 28 días (8, 9) y ha disminuido a lo largo del tiempo (4); el egreso de los pacientes puede darse a su domicilio en el 50% de los casos, y por fallecimiento en el 6 y el 63% (8, 10-16); los pacientes que sobreviven pueden presentar algún compromiso serio de su estado de salud.
En términos de discapacidad y sobrevida, del 25 al 69 % de los pacientes puede presentar un mal desenlace aun cuando se encuentren en un buen estado neurológico al momento del ictus (7, 13, 15-17); la mortalidad por este padecimiento puede ser de 6-63% (3, 8, 10-13, 15, 16). Este resultado está relacionado con la severidad inicial del padecimiento y con las complicaciones que se presenten durante su evolución.
Como consecuencia de la hemorragia, pueden presentarse edema, vasoespasmo cerebral tardío (VCT) e infarto cerebral además de hiperglucemia (1, 7, 8, 14, 16-18) (Fig. 1); estas complicaciones determinan, al menos en parte, el desenlace de los pacientes (7, 16, 17, 19). Por estas razones, muchos estudios se han centrado en el papel de la regulación de los niveles de glucosa en la evolución y el desenlace de la HSAa.

METABOLISMO CEREBRAL DE LA GLUCOSA EN LA HEMORRAGIA SUBARACNOIDEA ANEURISMÁTICA
En condiciones fisiológicas, el cerebro metaboliza la glucosa a través de la glucólisis acoplada al ciclo de Krebs y a la vía de las pentosas fosfato; prácticamente, toda la energía que utiliza el cerebro se obtiene a través del metabolismo de la glucosa (20). Los astrocitos contribuyen a mantener estos mecanismos almacenando glucógeno, aunque el cerebro contiene sólo una centésima parte del que está presente en el hígado, y solo podría mantener su funcionamiento por un máximo de 10 min (20). Estos procesos fisiológicos pueden verse afectados como consecuencia de una enfermedad neurológica.
A nivel cerebral, la HSAa causa un aumento del 30 al 50% en la captación de desoxiglucosa y una disminución en el flujo sanguíneo cerebral (FSC) (21-24).Utilizando técnicas de microdiálisis que permiten lograr un recobro cercano al 100%, la concentración de glucosa en el líquido intersticial cerebral de pacientes con HSAa puede ser de 0 a 5,5 mM y disminuye durante el curso de la enfermedad (25, 26); la concentración basal en estas condiciones se estima entre 1 y 4 mM (25). Los alcances y limitaciones de la microdiálisis intracerebral para la práctica clínica han sido discutidos previamente (20, 27, 28). Estos resultados podrían estar relacionados con la severidad de la enfermedad; de hecho, la concentración de glucosa es más baja en los pacientes que presentan déficit neurológico agudo respecto a quienes lo presentan de forma tardía o a quienes no lo presentan (29).
Otros marcadores también sugieren un cambio en el metabolismo de la glucosa en la HSAa. Tanto la concentración de lactato como el cociente lactato/piruvato son indicadores confiables del metabolismo anaerobio (27, 28). La HSAa provoca un aumento en el cociente lactato/piruvato en el tejido cerebral (30). La concentración de lactato medida por microdiálisis puede ser de 1 a 20 mM y la razón lactato/piruvato de 15 a 2.900; los niveles basales en estas condiciones se estiman entre 1 y 4 mM para lactato y entre 10 y 40 para el cociente lactato/piruvato (26, 31). Estos son algunos de los cambios en el metabolismo energético que se observan en la HSAa pero no son exclusivos de esta patología.
Puede detectarse un aumento en las concentraciones de lactato y piruvato, así como en el cociente lactato/piruvato en el líquido cefalorraquídeo (LCR) de pacientes con hemorragias subaracnoideas de diferentes etiologías respecto a sujetos control, al igual que en modelos experimentales de hemorragias subaracnoidea e intracerebral (32, 33). Este cambio del metabolismo aerobio al anaerobio refleja con mucha probabilidad los procesos de neurotoxicidad que provocan el déficit neurológico en la HSAa.
RELEVANCIA DEL METABOLISMO DE LA GLUCOSA PARA EL DESENLACE EN LA HEMORRAGIA SUBARACNOIDEA ANEURISMÁTICA
Al metabolismo de la glucosa se le ha relacionado con la evolución y el desenlace de la HSAa. El aumento en la concentración de lactato y de la razón lactato/piruvato, así como la disminución en la concentración de glucosa, ocurren de forma paralela a la presencia de déficit neurológico (26, 31), se asocian con un mal estado clínico y preceden el aumento en el flujo sanguíneo cerebral (FSC) relacionado con el vasoespasmo cerebral tardío (VCT), así como el déficit neurológico que acompaña (26, 34). El aumento en la concentración de lactato en el líquido cefalorraquídeo (LCR) está asociado con un peor desenlace de estos pacientes (32).
De este modo, los estudios indican que luego de la HSAa, ocurre una disminución del FSC y un aumento en el metabolismo anaerobio que disminuye la concentración de glucosa; desde el punto de vista teórico, puede sugerirse que esta menor disponibilidad junto con un mayor requerimiento metabólico a nivel central estimulan la liberación de glucosa desde órganos periféricos hacia el torrente sanguíneo, desde donde podría proveer al sistema nervioso central.
A nivel periférico, la manifestación es compatible, aunque aparentemente paradójica, con la que se observa en el sistema nervioso central pues, en contraste con la disminución observada en LCR y en líquido intersticial cerebral, se presenta un aumento en la concentración sérica de glucosa.
En la HSAa, pueden presentarse eventos hiperglucémicos aislados (glucemia> 200 mg/dL por menos de 48 horas) en el 36% de los casos, o persistentes (menores de 48 horas) en el 27% de ellos (9). Su tratamiento puede ser complicado puesto que puede mantenerse durante la hospitalización, en la tercera parte de los casos, a pesar de la intervención con insulina (9, 13); esto es de suma importancia pues guarda relación con la evolución y el desenlace de los pacientes (Fig. 1).
Es tal la relevancia de la hiperglucemia en esta enfermedad que incluso es un factor de riesgo para las secuelas neurológicas. La hiperglucemia de ingreso está asociada con un peor desenlace (RM 2,8; IC 95% 1,6-4,8) y una mayor mortalidad en la HSAa (RR 2,3; IC 95% 1,0-4,8) (10, 13, 35-38), aunque algunos estudios difieren de esos resultados (39). Un peor estado neurológico se relaciona con mayores niveles máximo y promedio de glucemia durante la hospitalización (40).
Estas alteraciones podrían estar relacionadas con el mecanismo de daño cerebral dado que el aumento en la concentración de lactato y de la razón lactato/piruvato, así como la disminución en la concentración de glucosa, ocurren de forma paralela a la presencia de déficit neurológico (26,31). El aumento en la concentración de lactato en el LCR está asociado con un peor desenlace de estos pacientes (32).
La interpretación de estos datos sobre hiperglucemia debe hacerse con cautela dado que el resultado opuesto muestra una asociación similar; en realidad, tanto la presencia de hiperglucemia (RM 2,8; IC 95% 1,2-6,5) (35, 39-41) como la de hipoglucemia (40) están asociadas con un mal desenlace, aunque existen más evidencias sobre la primera de ellas.
La hiperglucemia persistente predice un mal desenlace (GOS 1-3) a 10meses del evento vascular (RM 6,9; IC 95% 1,0-44,7) (9). Estos resultados pueden observarse en sangre venosa, mientras que la concentración arterial de glucosa no parece estar asociada con el desenlace (11); otros estudios muestran solo una asociación débil (12).
A partir de esto, puede sugerirse que la hiperglucemia se encuentra involucrada con los mecanismos fisiopatológicos que predisponen a un mal desenlace y, por tanto, que la intervención sobre ella podría prevenirlo. De hecho, la hiperglucemia de ingreso que es corregida durante la hospitalización ya no se asocia con un mal desenlace a dos semanas del ictus (9) e, incluso, reduce la probabilidad de presentarlo (RM 0,2; IC 95% 0,1-0,8) (13). Esto sugiere que la hiperglucemia se relaciona con las complicaciones que se presentan durante la evolución de la HSAa y determinan su desenlace; entre ellas, el VCT.
RELACIÓN ENTRE LA HIPERGLUCEMIA Y EL VASOESPASMO CEREBRAL TARDÍO
El vasoespasmo cerebral puede presentarse entre 4 y 12 días después del ictus (1) y en algunos casos genera síntomas neurológicos. Aproximadamente la mitad de los pacientes con vasoespasmo detectado por angiografía presentan síntomas relacionados (1). Puede presentarse vasoespasmo cerebral tardío (VCT) sintomático entre el 11 y el 44% de los casos, de 3 a 14 días después del ictus (7, 9, 12, 13, 42, 43); también pueden presentarse déficits neurológicos agudos en el 30%, o tardíos en el 25%, de los casos (31). Si bien el VCT es consecuencia de la hemorragia, el hecho de que se presenten alteraciones fisiológicas desde el inicio de la enfermedad mientras que el vasoespasmo es más tardío sugiere que este último es, más que secundario a la hemorragia, una consecuencia de las alteraciones que se presentan en los primeros días de la HSAa; entre ellas, la hiperglucemia.
El VCT está relacionado de alguna forma con el metabolismo de la glucosa. La glucemia promedio es mayor en los pacientes que desarrollan VCT sintomático, predice su aparición (RR 1,01; IC 95% 1,00-1,03) y está asociada con un mal desenlace (44). La concentración de glucosa al ingreso (44) o su máximo durante la hospitalización (42) no tiene ese valor predictivo. La presencia de hiperglucemia durante la estancia en la unidad de cuidados intensivos está asociada con una aparición más temprana del VCT (RR 1,9) (35), y precede al aumento en el FSC relacionado con el VCT, así como el déficit neurológico que lo acompaña (26, 34) (Fig. 1).
Puede sugerirse que este tipo de complicaciones de la HSAa fueran más severas o frecuentes en casos de hiperglucemia crónica, como en la diabetes, pero esto podría estar influenciado por el control que se tenga de la enfermedad; sin embargo, algunos estudios indican que un mal control de la glucemia (glucosa diaria promedio > 140 mg/dL) no está asociado con una mayor incidencia de VCT (12).
Por otro lado, algunos estudios muestran que la diabetes es predictora de VCT (RM 9,9) (12), aunque otros no lograron mostrar una asociación (44). En pacientes diabéticos con HSAa, la concentración promedio de glucosa sérica es de 130 mg/dL en el día en que se detecta el VCT, y puede aumentar más allá de los 150 mg/dL (250 mg/dL) en algunos pacientes) a los 6 días posteriores al VCT, aunque la tendencia de aumento es gradual desde el inicio de la HSAa, por lo que no parece que fuera consecuencia del VCT (12).
Algunos otros estudios sugieren que es más bien la hipoglucemia la que se relaciona con la aparición de vasoespasmo e infarto cerebrales en los pacientes con HSAa (40). De hecho, la disminución severa de glucosa por administración de insulina se asocia con un aumento en la concentración extracelular de glutamato y en el cociente lactato/piruvato a nivel cerebral (27), lo que podría potenciar el daño; adicionalmente, la hipoglucemia aumenta la mortalidad asociada a la HSAa en modelos experimentales (45) aunque algunos estudios clínicos no muestran ningún valor predictivo de la hipoglucemia para el VCT (35).
En cualquiera de los casos, el control de la hiperglucemia en la HSAa debe ser cuidadoso pues no puede descartarse que, en caso de ser extremo, resulte más bien contraproducente.
Complementarios a los estudios clínicos, los resultados experimentales no solo coinciden en que existe una relación entre la hiperglucemia y el VCT sino que además permiten sugerir el mecanismo que la explica. En modelos experimentales de diabetes se observa una disminución de la sensibilidad de la aorta al efecto vasodilatador de la acetilcolina o la histamina, sin alterarse el efecto vasoconstrictor de la norepinefrina (46-48). El aumento agudo en la concentración extracelular de glucosa (22 mM, aproximadamente 400 mg/dL) tiene un efecto similar (49), lo que sugiere que la hiperglucemia podría modular los mecanismos de vasorrelajación y vasoconstricción.
La hiperglucemia severa podría interferir con la relajación del músculo liso vascular al inhibir la síntesis de óxido nítrico (NO). La elevación de la concentración de glucosa a aproximadamente 460 mg/dL inhibe la actividad de la dimetilargininasa, la enzima que degrada a los inhibidores endógenos de la sintasa del NO (NOS), en células de músculo liso vascular por un mecanismo que involucra a alguna especie reactiva de oxígeno (50) (Fig. 2). Esto se refleja en que la glucosa, que a partir de 270 mg/dL, aumenta la concentración de dimetilarginina asimétrica (inhibidor endógeno de la sintasa del óxido nítrico, NOS) y disminuye la de GMPc (50) (Fig. 2).Esto contrasta con los estudios que sugieren un aumento en la síntesis del NO en la HSAa (51) que podría estar relacionado con el proceso inflamatorio que acompaña a la enfermedad; la discrepancia podría explicarse por la actividad de diferentes isoformas de la NOS, la del sistema vascular y la de la respuesta inflamatoria. En conjunto, algunos estudios indican que la glucosa puede reducir la vasorrelajación, otros estudios muestran que también puede estimular a moléculas vasoconstrictoras.
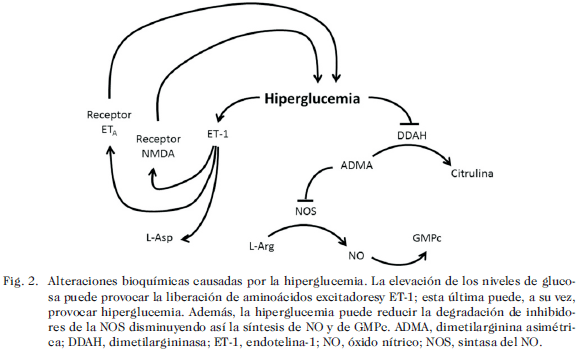
La hiperglucemia (glucosa 11,1 mM, aproximadamente 200 mg/dL) aumenta en 35% la secreción de endotelina-1 (ET-1) por las células endoteliales de la aorta a través de un mecanismo independiente del cambio en la presión osmótica (52) (Fig. 2).Además del vasoespasmo, esto puede favorecer la isquemia cerebral dado que la ET-1 provoca la liberación de un aminoácido excitador como aspartato en células granulares del cerebelo por un mecanismo que depende de la concentración extracelular de calcio (53) (Fig. 2). Esto, entre otros mecanismos, sugiere que la hiperglucemia podría inducir VCT e isquemia, aunque la modulación entre estos mecanismos puede ser recíproca.
Algunos otros estudios avalan la interacción entre la ET-1 y los aminoácidos excitadores. La administración intracerebroventricular de ET-1 aumenta la concentración de glucosa en sangre por un mecanismo dependiente de los receptores ETA y NMDA (54, 55) (Fig. 2). Su administración intra-arterial también tiene un efecto hiperglucemiante (56). Adicionalmente, la administración i.c.v. de ET-1 aumenta la actividad metabólica cerebral medida por la captación de desoxiglucosa, como se observa en la HSAa, a través de los receptores ETA y NMDA (54, 55).
Por estas razones, puede concluirse que la relación entre la hiperglucemia y el VCT es recíproca y cualquiera de las dos alteraciones podría ser la causa de la otra, pero la temporalidad de los eventos en el escenario clínico indica más bien que la hiperglucemia precede a la aparición del VCT (Fig. 1).
FACTORES DE RIESGO Y PREVENCIÓN DEL VASOESPASMO CEREBRAL TARDÍO
Entre los principales factores de riesgo de desarrollo de VCT que se consideran en la práctica clínica se encuentran algunas de las características que pueden observarse desde el momento del ictus. De hecho, se considera que el principal predictor de vasoespasmo es la cantidad de sangre presente en el espacio subaracnoideo en la tomografía axial computada inicial clasificada a través de la escala de Fisher original y modificada (1, 43). Otros factores de riesgo relacionados con el sistema vascular son la elevación de la presión arterial media más allá de 112 mmHg al ingreso e historia de hipertensión arterial (43). Algunos otros, que se relacionan más con las funciones neurológicas que (aparentemente) con el sistema vascular son, por ejemplo, la escala de la Federación Mundial de Cirujanos Neurológicos (WFNS) y la edad menor a 50 años (5, 43). Sin embargo, los análisis mutivariados han mostrado que solamente la escala de Fisher modificada es un predictor independiente de VCT (43). Sin embargo, la remoción del hematoma no reduce la incidencia de VCT (43), lo que sugiere que esta complicación es causada por otros mecanismos. Con esto, el mecanismo la relación entre la magnitud de la hemorragia y la hiperglucemia que ocurre desde el ictus puede cobrar gran relevancia en términos de etiopatogenia.
A partir de estos factores de riesgo puede iniciarse una intervención para prevenir el VCT, aunque no se cuenta con una que sea del todo eficaz (57). Se ha descrito que el tratamiento temprano (ya sea quirúrgico o endovascular) puede prevenir el VCT; también se utilizan los bloqueadores de canales de calcio y la terapia triple H (hipertensión, hipervolemia y hemodilución) (57, 58). A pesar de estos esfuerzos, el VCT sigue siendo una causa importante de morbimortalidad en la HSAa.
EFECTO DE LA HIPERGLUCEMIA SOBRE EL EDEMA CEREBRAL
El edema cerebral puede estar presente desde el ingreso a hospitalización en el 8% de los pacientes con hemorragia subaracnoidea no traumática mientras que el 12% puede desarrollarlo, en un promedio de 6 días después del ictus (1). Se presenta durante el curso de la enfermedad, lo que sugiere que es provocado por alguno de los mecanismos fisiopatológicos que ocurren desde que ésta inicia como, puede ser, la hiperglucemia.
A nivel experimental, la hiperglucemia (400 mg/dL) aumenta discretamente (4%) la magnitud del edema secundario a la hemorragia intracerebral (59). Este efecto podría estar relacionado con la alteración de los niveles de iones luego de la isquemia dado que la hiperglucemia (de aproximadamente 400 mg/dL) provoca un aumento en el contenido cerebral de Na+ (56%) y de Ca2+ (70%) así como una disminución en el de K+ (18%) respecto de la normoglucemia, luego de la isquemia cerebral (60).
Los estudios clínicos no coinciden por completo con esos resultados, puesto que la concentración máxima de glucosa durante la hospitalización no está asociada con la magnitud de edema cerebral al ingreso de los pacientes con HSAa (38), lo que podría explicarse por el nivel tan elevado de glucosa que debe alcanzarse a nivel experimental para observar su efecto, de modo que determinar la relevancia de la hiperglucemia para el edema cerebral requiere estudios adicionales.
Por todo lo anterior, debe considerarse que el control de la glucemia podría jugar un papel central en la evolución y el desenlace de la HSAa. Se han descrito los mecanismos a través de los cuales puede darse esta relación, los cuales incluyen el equilibrio de iones, la liberación de aminoácidos excitadores, la estimulación de moléculas vasoconstrictoras y la disminución en la síntesis de vasorrelajantes. Sin embargo, existen estudios que no apoyan la hipótesis sobre la participación de la hiperglucemia en esta enfermedad. En conjunto, estas evidencias sugieren que el control de los niveles de glucosa podría modificar el desenlace de los grupos de pacientes con HSAa dependiendo de las complicaciones que presenten.
REFERENCIAS
1. Fugate JE, Rabinstein AA. Intensive care unit management of aneurysmal subarachnoid hemorrhage. Curr Neurol Neurosci Rep 2012; 12: 1-9. [ Links ]
2. de Rooij NK, Linn FHH, van der Plas JA, Algra A, Rinkel GJE. Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry 2007; 78:1365-1372. [ Links ]
3. Nieuwkamp DJ, Setz LE, Algra A, Linn FH, de Rooij NK, Rinkel GJ. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol 2009; 8: 635-642. [ Links ]
4. Rincon F, Rossenwasser RH, Dumont A. The epidemiology of admissions of nontraumatic subarachnoid hemorrhage in the United States. Neurosurgery 2013; 73: 217-222. [ Links ]
5. Rabb CH, Tang G, Chin LS, Giannotta SL. A statistical analysis of factors related to symptomatic cerebral vasospasm. Acta Neurochir (Wien) 1994; 127:27-31. [ Links ]
6. Schwartz JH. Circulación arterial del cerebro. En: Kandel ER, Schwartz JH, Jessell TM, editores. Principios de Neurociencia. 4ª Ed. Madrid: McGraw Hill; 2000. [ Links ]
7. Bargés-Coll J, Pérez-Neri I, Avendaño J, Méndez-Rosito D, Gómez-Amador JL, Ríos C. Plasma taurine as a predictor of poor outcome in patients with mild neurological deficits after aneurysmal subarachnoid hemorrhage. J Neurosurg 2013; 119: 1021-1027. [ Links ]
8. Navi BB, Kamel H, McCulloch CE, Nakagawa K, Naravetla B, Moheet AM, Wong C, Johnston SC, Hemphill JC 3rd, Smith WS. Accuracy of neurovascular fellows’ prognostication of outcome after subarachnoid hemorrhage. Stroke 2012; 43: 702-707. [ Links ]
9. McGirt MJ, Woodworth GF, Ali M, Than KD, Tamargo RJ, Clatterbuck RE. Persistent perioperative hyperglycemia as an independent predictor of poor outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg 2007; 107: 1080-1085. [ Links ]
10. Alberti O, Becker R, Benes L, Wallenfang T, Bertalanffy H. Initial hyperglycemia as an indicator of severity of the ictus in poor-grade patients with spontaneous subarachnoid hemorrhage. Clin Neurol Neurosurg 2000; 102: 78-83. [ Links ]
11. Barcelos GK, Tholance Y, Grousson S, Renaud B, Perret-Liaudet A, Dailler F, Zimmer L. Outcome of poor-grade subarachnoid hemorrhage as determined by biomarkers of glucose cerebral metabolism. Neurocrit Care 2013; 18: 234-244. [ Links ]
12. Dumont T, Rughani A, Silver J, Tranmer BI. Diabetes mellitus increases risk of vasospasm following aneurysmal subarachnoid hemorrhage independent of glycemic control. Neurocrit Care 2009; 11: 183-189. [ Links ]
13. Latorre JG, Chou SH, Nogueira RG, Singhal AB, Carter BS, Ogilvy CS, Rordorf GA. Effective glycemic control with aggressive hyperglycemia management is associated with improved outcome in aneurysmal subarachnoid hemorrhage. Stroke 2009; 40: 1644-1652. [ Links ]
14. Oertel MF, Schwedler M, Stein M, Wachter D, Scharbrodt W, Schmidinger A, Böker DK. Cerebral energy failure after subarachnoid hemorrhage: the role of relative hyperglycolysis. J ClinNeurosci 2007; 14: 948-954. [ Links ]
15. Vergouwen MDI, de Haan RJ, Vermeulen M, Roos YBWEM. Effect of statin treatment on vasospasm, delayed cerebral ischemia, and functional outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis update. Stroke 2010; 41: e47-e52. [ Links ]
16. Vergouwen MDI, Ilodigwe D, Macdonald RL. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke 2011; 42: 924-929. [ Links ]
17. Pérez-Neri I, Bargés-Coll J, Ramírez-Bermúdez J, García-López R, Ojeda-López C, Méndez-Rosito D, Ramírez-Abascal M, Márquez-Flores MA, Nente F, Avendaño J, Montes S, Gómez-Amador JL, Moreno-Jiménez S, Soto-Hernández JL, Ríos C. Taurina es un marcador de pronóstico, complicaciones y supervivencia de pacientes con diferentes trastornos neurológicos. Arch Neurocien (Mex) 2011; 16(Supl II): 22-24. [ Links ]
18. Liu Y, Soppi V, Mustonen T, Könönen M, Koivisto T, Koskela A, Rinne J, Vanninen RL. Subarachnoid hemorrhage in the subacute stage: elevated apparent diffusion coefficient in normal-appearing brain tissue after treatment. Radiology 2007; 242: 518-525. [ Links ]
19. Fergusen S, Macdonald RL. Predictors of cerebral infarction in patients with aneurysmal subarachnoid hemorrhage. Neurosurgery 2007; 60: 658-667. [ Links ]
20. Castelazo-Arredondo LA, Castelazo-Aguirre M. Monitoreo metabólico cerebral. En: Carrillo Esper R, Castelazo Arredondo A, eds. Neuromonitoreo en medicina intensiva y anestesiología. México: Alfil; 2011. [ Links ]
21. Delgado TJ, Arbab MAR, Diemer NH, Svendgaard NA. Subarachnoid hemorrhage in the rat: cerebral blood flow and glucose metabolism during the late phase of cerebral vasospasm. J Cereb Blood Flow Metab 1986; 6: 590-599. [ Links ]
22. Delgado-Zygmunt T, Arbab MAR, Shiokawa Y, Svendgaard NA. Cerebral blood flow and glucose metabolism in the squirrel monkey during the late phase of cerebral vasospasm. Acta Neurochir (Wien) 1993; 121: 166-173. [ Links ]
23. Svendgaard NA, Goksel M, Westring S. Trigeminal nerve and brainstem catecholamine systems in cerebral vasospasm. Neurol Med Chir Suppl (Tokyo) 1998; 38: 146-151. [ Links ]
24. Yamamoto S, Teng W, Nishizawa S, Kakiuchi T, Tsukada H. Improvement in cerebral blood flow and metabolism following subarachnoid hemorrhage in response to prophylactic administration of the hydroxyl radical scavenger, AVS, (±)-N,N’-propylenedinicotinamide: a positron emission tomography study in rats. J Neurosurg 2000; 92: 1009-1015. [ Links ]
25. Hoelper BM, Hofmann E, Sporleder R, Soldner F, Behr R. Transluminal balloon angioplasty improves brain tissue oxygenation and metabolism in severe vasospasm after aneurysmal subarachnoid hemorrhage: case report. Neurosurgery 2003; 52: 970-976. [ Links ]
26. Nilsson OG, Brandt L, Ungerstedt U, Säveland H. Bedside detection on brain ischemia using intracerebral microdialysis: subarachnoid hemorrhage and delayed ischemic deterioration. Neurosurgery 1999; 45: 1176-1184. [ Links ]
27. Goodman JC, Robertson CS. Microdialysis: is it ready for prime time? Curr Opin Crit Care 2009; 15: 110-117. [ Links ]
28. Ramírez-Segura EH, León-Álvarez E. Microdiálisis: una herramienta más de monitoreo cerebral. En: Carrillo Esper R, Castelazo Arredondo A, eds. Neuromonitoreo en medicina intensiva y anestesiología. México: Alfil; 2011. [ Links ]
29. Sarrafzadeh AS, Sakowitz OW, Kiening KL, Benndorf G, Lanksch WR, Unterberg AW. Bedside microdialysis: a tool to monitor cerebral metabolism in subarachnoid hemorrhage patients? Crit Care Med 2002; 30: 1062-1070. [ Links ]
30. Fein JM. Cerebral energy metabolism after subarachnoid hemorrhage. Stroke 1975; 6: 1-8. [ Links ]
31. Unterberg AW, Sakowitz OW, Sarrafzadeh AS, Benndorf G, Lanksch WR. Role of bedside microdialysis in the disgnosis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 2001; 94: 740-749. [ Links ]
32. Fujishima M, Sugi T, Choki J. Cerebrospinal fluid and arterial lactate, pyruvate and acid-base balance in patients with intracranial hemorrhages. Stroke 1975; 6: 707-774. [ Links ]
33. Sugi T, Fujishima M, Omae T. Lactate and pyruvate concentrations, and acid-base balance of cerebrospinal fluid in experimentally induced intracerebral and subarachnoid hemorrhage in dogs. Stroke 1975; 6: 715-719. [ Links ]
34. Skjøth-Rasmussen J, Schulz M, Kristensen SR, Bjerre P. Delayed neurological deficits detected by an ischemic pattern in the extracellular cerebral metabolites in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg 2004; 100: 8-15. [ Links ]
35. Charpentier C, Audibert G, Guillemin F, Civit T, Ducrocq X, Bracard S, Hepner H, Picard L, Laxenaire MC. Multivariate analysis of predictors of cerebral vasospasm occurrence after aneurysmal subarachnoid hemorrhage. Stroke 1999; 30: 1402-1408. [ Links ]
36. Claassen J, Vu A, Kreiter KT, Kowalski RG, Du EY, Ostapkovich N, Fitzsimmons BF, Connolly ES, Mayer SA. Effect of acute physiologic derangements on outcome after subarachnoid hemorrhage. Crit Care Med 2004; 32: 832-838. [ Links ]
37. DorhoutMees SM, van Dijk GW, Algra A, Kempink DRJ, Rinkel JGE. Glucose levels and outcome after subarachnoid hemorrhage. Neurology 2003; 61: 1132-1133. [ Links ]
38. Yoshimoto Y, Tanaka Y, Hoya K. Acute systemic inflammatory response syndrome in subarachnoid hemorrhage. Stroke 2001; 32: 1989-1993. [ Links ]
39. Frontera JA, Fernandez A, Claassen J, Schmidt M, Schumacher HC, Wartenberg K, Temes R, Parra A, Ostapkovich ND, Mayer SA. Hyperglycemia after SAH: predictors, associated complications, and impact on outcome. Stroke 2006; 37: 199-203. [ Links ]
40. Naidech AM, Levasseur K, Liebling S, Garg RK, Shapiro M, Ault ML, Afifi S, Batjer HH. Moderate hypoglycemia is associated with vasospasm, cerebral infarction, and 3-month disability after subarachnoid hemorrhage. Neurocrit Care 2010; 12: 181-187. [ Links ]
41. Tseng MY, Hutchinson PJ, Turner CL, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Biological effects of acute pravastatin treatment in patients after aneurysmal subarachnoid hemorrhage: a double-blind, placebo-controlled trial. J Neurosurg 2007; 107: 1092-1100. [ Links ]
42. Yahia AM, Kirmani JF, Qureshi AI, Guterman LR, Hopkins LN. The safety and feasibility of continuous intravenous magnesium sulfate for prevention of cerebral vasospasm in aneurysmal subarachnoid hemorrhage. Neurocrit Care 2005; 3: 6-23. [ Links ]
43. Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ES Jr, MacDonald RL, Mayer SA. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59: 21-27. [ Links ]
44. Badjatia N, Topcuoglu MA, Buonanno FS, Smith EE, Nogueira RG, Rordorf GA, Carter BS, Ogilvy CS, Singhal AB. Relationship between hyperglycemia and symptomatic vasospasm after subarachnoid hemorrhage. Crit Care Med 2005; 33: 1603-1609. [ Links ]
45. Thal SC, Engelhard K, Werner C. New cerebral protection strategies. Curr Opin Anaesthesiol 2005; 18: 490-495. [ Links ]
46. Kamata K, Miyata N, Kasuya Y. Impairment of endothelium-dependent relaxation and changes in levels of cyclic GMP in aorta from streptozotocin-induced diabetic rats. Br J Pharmacol 1989; 97: 614-618. [ Links ]
47. Meraji S, Jayakody L, Senaratne MPJ, Thomson ABR, Kappagoda T. Endothelium-dependent relaxation in aorta of BB rat. Diabetes 1987; 36: 978-981. [ Links ]
48. Oyama Y, Kawasaki H, Hattori Y, Kanno M. Attenuation of endothelium-dependent relaxation in aorta from diabetic rats. Eur J Pharmacol 1986; 131: 75-78. [ Links ]
49. Tesfamanam B, Brown ML, Cohen RA. Elevated glucose impairs endothelium-dependent relaxation by activating protein kinase C. J Clin Invest 1991; 87: 1643-1648. [ Links ]
50. Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus role of asymmetric dimethylarginine and dimethylarginine dimethylamino hydrolase. Circulation 2002; 106:987-992. [ Links ]
51. Pérez-Neri I, Castro E, Montes S, Boll MC, Bargés-Coll J, Soto-Hernández JL, Ríos C. Arginine, citrulline and nitrate concentrations in the cerebrospinal fluid from patients with acute hydrocephalus. J Chromatogr B 2007; 851: 250-256. [ Links ]
52. Yamauchi T, Ohnaka K, Takayanagi R, Umeda F, Nawata H. Enhanced secretion of endothelin-1 by elevated glucose levels from cultured bovine aortic endothelial cells. FEBS Lett 1990; 267: 16-18. [ Links ]
53. Chuang DM, Lin WW, Lee CY. Endothelium-induced activation of phosphoinositide turnover, calcium movilization, and transmitter release in cultured neurons and neurally related cell types. J Cardiovasc Pharmacol 1991; 17(Suppl 7):S85-S88. [ Links ]
54. Chew BH, Weaver DF, Balaban CD, Gross PM. NMDA-mediated metabolic activation of the cerebellar cortex in behaving rats by the neuropeptide endothelin-1. Brain Res 1994; 647: 345-352. [ Links ]
55. Gross PM, Weaver DF, Ho LT, Pang JJ, Envinsson L. FR139317, a specific ETA-receptor antagonist, inhibits cerebral activation by intraventricular endothelin-1 in conscious rats. Neuropharmacology 1994; 33: 1155-1166. [ Links ]
56. Willette RN, Sauermelch C, Ezekiel M, Feuerstein G, Ohlstein OH. Effect of endothelin on cortical microvascular perfusion in rats. Stroke 1990; 21: 451-458. [ Links ]
57. Oyama K, Criddle L. Vasospasm after aneurysmal subarachnoid hemorrhage. Crit Care Nurse 2004; 24: 58-67. [ Links ]
58. van Gijn J, Kerr RS, Rinkel GJE. Subarachnoid haemorrhage. Lancet 2007; 369: 306-318. [ Links ]
59. Song EC, Chu K, Jeong SW, Jung KH, Kim SH, Kim M, Yoon BW. Hyperglycemia exacerbates brain edema and perihematomal cell death after intracerebral hemorrhage. Stroke 2003; 34: 2215-2220. [ Links ]
60. Warner DS, Smith ML, Siesjö BK. Ischemia in normo- and hyperglycemic rats: effects on brain water and electrolytes. Stroke 1987; 18: 464-471. [ Links ]
