










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos Venezolanos de Farmacología y Terapéutica
versión impresa ISSN 0798-0264
AVFT v.23 n.2 Caracas jul. 2004
Helicobacter Pylori: Enteropatógeno frecuente del ser humano
M Rivera1, F Contreras2, A Terán3 y C Fouillioux4.
1 Médico internista Gastroenterólogo, profesor asistente Cátedra de Fisiopatología, Medicina, UCV.
2 Médico internista, profesor agregado Cátedra de Fisiopatología, Medicina, UCV.
3 Residente Posgrado, Hospital Victorino Santaella, Los Teques, Edo. Miranda
4 Prof. Bioquímica, EEE - UCV
E-mail: mrivera@cantv.net
RESUMEN
En este artículo se revisan los conceptos recientes en relación a una infección bacteriana cuyo agente causal es Helicobacter Pylori, frecuentemente implicado en cuadros gastrointestinales diversos y responsable directo de la enfermedad úlcero-péptica que además es reconocido como agente carcinógeno tipo I por su asociación con cáncer gástrico. Helicobacter Pylori es un bacilo gram negativo que coloniza la mucosa gástrica de amplia distribución mundial, presentes con una alta frecuencia en pacientes que consultan con sintomatología gastrointestinal, por lo que se analizan aspectos básicos de su fisiología, sus diferentes morfologías que directamente van a repercutir en su patogenicidad y se detallan los mecanismos fisiopatológicos responsables de los diferentes cuadros clínicos, los métodos endoscópicos y de laboratorio específicos para el diagnóstico y los avances más recientes en el tratamiento y profilaxis de la enfermedad.
Palabras Clave: Helicobacter Pylori, Enfermedad úlcereo-péptica, Diagnóstico y tratamiento.
ABSTRACT
This paper reviews current knowledge in relation to the bacterial infection caused by Helicobacter pylori, frequently associated to gastrointestinal diseases, and direct cause of peptic ulcer disease. Besides this, it is recognized as a carcinogenic agent, given its association with gastric carcinoma. Helicobacter pylori, is a gram negative bacillus that colonizes gastric mucosa, with a worldwide distribution, and its highly detected in patients with gastrointestinal symptoms, therefore its physiology and different morphologies that exert an effect over its pathogenicity are described, among the physiopathologic mechanisms underlying clinical presentations, endoscopic methods and biochemistry tests specific for diagnosis and finally treatment.
Key Words: Pylori helicobacter, Peptic ulcer disease, Diagnostic of treatment.
INTRODUCCIÓN
Helicobacter pylory (H pylori) fue identificada por los investigadores australianos Barry Marshall y Robin Warren en 1982. En sus estudios, todos los pacientes que tenían úlceras duodenales y el 80% de los pacientes que tenían úlceras gástricas tenían la bacteria. Previamente, a finales del siglo XIX Bizzozero había descrito la presencia de bacterias espirales en el estómago de perros y gatos, lo que abrió sin dudas las puertas a la investigación. Desde 1989 H pylori se considera la especie tipo de un nuevo género Helicobacter en el que existen al menos otras 19 especies y que es responsable directo de la úlcera péptica(1).
Helicobacter Pylori (H pylori) es un bacilo Gram negativo que coloniza la mucosa gástrica en el 90-95% de los pacientes con úlcera duodenal y en el 60-70% de los pacientes con úlcera gástrica. Habita en la capa que reviste las células epiteliales y en menor grado adherido a ellas. Antes conocido como Campylobacter pyloridis o Campylobacter pylori por su aspecto en la tinción de Gram y su requerimiento microaaerofílico aunque presentaba ciertas características atípicas; sin embargo, los estudios genómicos modernos, especialmente el análisis de secuencias del ácido ribonucleico ribosomal 16S (ARNr) permitieron demostrar que Campylobacter y Helicobacter eran dos géneros diferentes(1).
La prevalencia de la infección por H pylori se ha estimado en 80 a 90% en los países en desarrollo. Generalmente la adquisición ocurre cerca de un 10% de niños por año, por consiguiente, la mayoría se encuentra infectado en la adolescencia(2,3,4). La situación en Venezuela no es diferente con respecto al resto de los países en desarrollo, habiéndose detectado su presencia en un 95 a 100% de las úlceras duodenales y en un 70 a 80% de las úlceras gástricas. En el estado Táchira existe una de la más alta prevalencia de infección (90 a 96%) y es allí donde también se reporta alta incidencia de carcinoma gástrico que se ha asociado a la presencia de H pylori en numerosos estudios(5).
La forma de transmisión no se conoce con exactitud. Hay datos que hacen pensar que se transmite de persona a persona y que el contagio es mayor cuanto mayor sea la proximidad y el contacto; en este sentido, el contagio en la escuela no parece importante y si lo es el ambiente familiar. Se ha aislado H pylori en la placa dental y en la saliva. Se ha investigado H pylori en el agua y los alimentos con resultados casi siempre negativos. El hallazgo de fragmentos de ADN de H pylori en ejemplares de la mosca doméstica ha hecho pensar en la posibilidad de su papel como agente de contagio(3).
TAXONOMÍA DE LA BACTERIA
Desde su cultivo en 1982 ha recibido diferentes denominaciones hasta adquirir el nombre definitivo:
CLO (Campylobacter like organism).
GCLO (Gastric Campylobacter like organism).
Campylobacter pyloridis.
Campylobacter pyloric.
Campylobacter pylori.
Helicobacter pylori: (1989) especie tipo de un nuevo género, Helicobacter.
Helicobacter, Campylobacter, Arcobacter y Wolinella pertenecen a un grupo distinto de bacterias (Superfamilia VI del ARNr) que está relacionado lejanamente con otras eubacterias.
CARACTERÍSTICAS DE HELICOBACTER PYLORI
H pylori es un bacilo multiflagelado gram negativo y microaerofílico que habita en la capa de moco del estómago, donde está parcialmente protegida del ácido clorhídrico. H pylori también se ha presentado en una forma cocoide viable pero no cultivable. Se especula que la forma cocoide sea una forma de resistencia capaz de soportar las condiciones adversas que encuentra H pylori en el medio ambiente, reversible a la forma espiral en el momento en que vuelvan a darse las condiciones óptimas(6).
H pylori es considerado generalmente como un microorganismo extracelular pero en varios estudios se ha encontrado en el interior de las células epiteliales; una invasión similar a la Yersinia Enterocolítica lo que sugiere que un mecanismo invasivo puede ser el responsable del daño a las células epiteliales y la ulceración péptica(7).
H pylori segrega ciertas proteínas que atraen a los macrófagos y neutrófilos produciendo inflamación en la zona afectada; produce además grandes cantidades de ureasa, la cual al hidrolizar la urea neutraliza el ácido del estómago en su entorno, mecanismo por el cual se protege aun más del medio externo. La bacteria segrega además proteasas, citotoxinas como interleukinas (IL)(8,9), factor de necrosis tumoral alfa (TNF alpha), factor de activación plaquetaria (PAF), interferón gamma (INF gamma), especies reactivas de oxígeno lipopolisacáridos y fosfolipasas que son las principales responsables del daño de la mucosa que genera el H pylori.
Todos los sucesos que ocurren en la mucosa gástrica van a depender de los factores de virulencia y de los mecanismos patógenos propios de la bacteria. El primero de ellos le va a permitir sobrevivir en el medio ácido hostil del estómago y los segundos son los que indirectamente le van a permitir producir el daño de la barrera mucosal gástrica.
Dentro de los factores de virulencia se encuentran: 1) La forma y los movimientos espirales. 2) Enzimas y proteínas de adaptación (ureasas, catalasas, proteína inhibidora de la secreción de ácido gástrico. 3) Habilidad para adherirse a las células de la mucosa gástrica y al moco (adhesinas bacterianas y receptores para células epiteliales).
Los mecanismos patógenos principalmente implicados son: 1) Producción de toxinas (citotoxinas, ureasas, mucinasa, lipasa, lipolisacaridasa, hemolisinas, Fosfolipasa A). 2) Mediadores de inflamación (activación de neutrófilos, activación de monocitos y macrófagos, leucotrienos, fenómenos autoinmunes, infiltración y degranulación de eosinófilos) y 3) Capacidad de contribuir con el incremento de la actividad ácido-péptica gástrica(10).
H pylori es muy móvil y activo, lo cual es resultado de su forma espiral y de los flagelos unipolares que le confiere una gran movilidad y le permite llegar a la mucosa y no ser eliminado por los mecanismos defensivos del huésped. Actualmente se ha sugerido que el flagelo es determinante como sensor de cambios de pH entre la luz gástrica y la capa de moco. La ureasa es producida en cantidades muy altas por H pylori, es esencial para su sobrevida, puesto que bacterias mutantes ureasa-deficientes no logran colonizar el estómago. La actividad de la ureasa permite el desdoblamiento rápido y permanente de la urea en el jugo gástrico, moco y mucosa, a substratos principales en la producción de amonio; aparentemente estos procesos proveen a la bacteria de la habilidad de exportar iones de hidrógeno desde dentro del citoplasma, regulando así el pH, y también crea un microambiente alcalino que lo protege de la acidez del estómago. Además de la ureasa, también produce superóxido-dismutasa y otras moléculas diversas que se encuentran involucradas en la adhesión específica a las células epiteliales del estómago. El H pylori posee gran variedad de adhesinas que reconocen de forma específica a los receptores de la mucosa gástrica y se unen a ellos comenzando la colonización bacteriana(11).
Las adhesinas del H pylori están constituidas por una hemaglutinina radiante desde la superficie de la bacteria con estructura de tipo afrimbial y un diámetro de 2 nm y perteneciente al grupo de los sialoconjugados. Es detectable utilizando eritrocitos de una variedad de especies con las propiedades de una sustancia antigénica termolábil y sensible a la acción de la pronasa, la papaína y la neuraminidasa; resistente a la pepsina y parcialmente a la tripsina. En general, la adhesión es ventajosa para la supervivencia del patógeno y para favorecer la liberación de las toxinas de los gérmenes directamente sobre las células epiteliales(11,12).
Junto a los factores antes mencionados, los cuales son producidos por todas las cepas de H pylori, existen algunos otros que son producidos por un subgrupo de bacterias aisladas. Clínicamente estas son proteínas asociadas a Citotoxina (CagA) y la Citotoxina Vacuolizante (VacA). La proteína VacA forma vacuolas en células epiteliales aisladas y ulceraciones superficiales en estómagos de animales de estudios(13).
Según análisis de expresión de estas proteínas y sus genes, H pylori puede ser clasificado en dos tipos: el tipo I donde la bacteria tiene el gen que codifica para CagA y expresa la proteína CagA y la citotoxina vacuolizante. El tipo II donde la bacteria no tiene el gen que codifica para el CagA y no expresa por consiguiente la proteína CagA o la citotoxina vacuolizante. El tipo I y II representa aproximadamente el 56 y 16% respectivamente, y el restante posee un fenotipo intermedio que expresa CagA independientemente de VacA o viceversa lo que hace pensar que CagA no es necesario para la expresión de la citotoxina vacuolizante(14,15).
La presencia del gen CagA y la expresión de su proteína, son reportados significativamente más altos en pacientes con úlcera duodenal y gastritis severa, incluyendo también gastritis atrófica y gastritis superficial. Recientemente dos genes asociados cercanamente han sido identificados y a su inicio se llamaron CagB y CagC, posteriormente fueron renombrados como PicB y PicC, siendo la terminología Pic referida a la habilidad de promover la inducción de citoquinas en el huesped; este clauster de nuevos genes podrían definir una importante región del genoma relacionada con la patogenicidad de la bacteria y podría llegar a representar la diferencia entre cepas patógenas y cepas comensales de H pylori(16). El conocimiento del genoma de H pylori ha permitido comprobar que dentro de la misma persona y al mismo tiempo hay muchos diferentes tipos de Helicobacter que compiten unos con otros para ocupar el mismo nicho. En esa competencia se desarrollan cambios tanto intragenómicos (dentro del mismo gérmen) como intergenómicos (entre gérmenes distintos)(17).
La infección por cepas de H pylori con el gen de antígeno A asociado a citotoxina (CagA) está asociada con el carcinoma gástrico. En Taiwan, H pylori con vacA s1a, cagA y iceA1 están presentes en la mayoría de los pacientes con cáncer gástrico y gastritis crónica(18,19). Se ha demostrado que VacA interrumpe la maduración de los fagosomas en los macrófagos como también modula e interfiere con las funciones inmunológicas de los linfocitos T; adicionalmente, las células infectadas por H pylori expresan receptores A y B para interleukina 8 lo cual puede contribuir al aumento de la respuesta inflamatoria. Numerosas evidencias relacionan la variación genética de la respuesta inflamatoria producida por H pylori con el mayor riesgo de carcinoma gástrico(16,20).
La infección por H pylori induce una respuesta inmunológica humoral sistémica y a nivel de la mucosa gástrica. La producción de anticuerpos no conduce a la erradicación de la infección pero puede contribuir al daño tisular. Algunos pacientes infectados tienen autoanticuerpos directamente contra la ATPasa de las células parietales lo cual se correlaciona con incremento de la atrofia del cuerpo gástrico(18).
En adición al daño asociado con traslocación de proteínas mediada a cag-PAI, la injuria epitelial de H pylori puede ser secundaria a otros mecanismos como liberación de especies de oxígeno y nitrógeno reactivos producidos por la activación de neutrófilos. La inflamación crónica también incrementa el recambio de células epiteliales y la apoptosis(21,22).
Después de la ingestión, H pylori coloniza predominantemente el antro gástrico, facilitado por los factores de virulencia y de patogenicidad antes mencionados, una vez allí, la infección persiste por años y tal vez de por vida, desencadenando y manteniendo una marcada respuesta inflamatoria que produce daños sobre la mucosa gástrica (gastritis superficial a gastritis crónica atrófica), además de producir alteración en los mecanismos de regulación de la secreción ácida gástrica por inhibición de la relación entre somatostatina y gastrina, lo cual trae como consecuencia, cambios de la motilidad antro-piloro-duodenal, pudiendo ocasionar síntomas, aunque un gran número de individuos se mantiene asintomático(23).
La gastrinemia basal aumenta aproximadamente en un 50% y la postprandial en un 100% en los pacientes en los pacientes infectados. De algún modo H pylori altera los mecanismos reguladores antrales, propiciando la producción inapropiada de gastrina. Además de la hipótesis de que esta alteración sea resultado de la actividad de la ureasa podría deberse también a una alteración de la liberación de gastrina inducida por los mecanismos que causan la gastritis antral. La alteración estructural que H pylori en la mucosa gástrica puede potenciar los mecanismos ulcerogénicos como la inhibición de la somatostatina. Una tercera posibilidad sería que la hipergastrinemia sea secundaria a una alteración de los mecanismos locales que regulan las células neuroendocrinas G y D. El rápido descenso de los niveles de gastrina como respuesta al tratamiento de erradicación se correlaciona mejor con una alteración de la regulación entre las células G y D que con un cambio en el número de estas(24).
CUADROS CLÍNICOS ASOCIADOS A LA INFECCIÓN POR HELICOBACTER PYLORI
El H pylori fue inicialmente observado en pacientes con gastritis, pero su descubrimiento se ha asociado, no solo con esta afección, sino también con úlcera péptica, linfomas y adenocarcinomas gástricos. En los países en desarrollo se estiman cifras de contaminación que resultan alarmantes. La vía de contaminación más probable es la oral y se le atribuye un papel fundamental a las aguas de consumo contaminadas.
La presencia de H pylori en el aparato gastrointestinal se acompaña invariablemente de manifestaciones de gastritis y en general las terapias antibióticas para erradicar el microorganismo son hoy en día el procedimiento eficaz en la terapéutica de la úlcera gastroduodenal(25,26). La asociación entre la infestación con H pylori y cáncer gástrico es tan estrecha que esta bacteria ha sido clasificada como carcinógeno de clase I, por la Agencia Internacional de Investigaciones sobre el cáncer y la OMS en 1994(27).
Las personas infectadas aumentan el riesgo de sufrir de adenocarcinoma gástrico y linfoma a lo largo de su vida entre 3 a 6 veces el riesgo normal, pasando de cifras de un 0,5% hasta un 1,5-3%.
H pylori se ha encontrado frecuentemente en pacientes con gastritis atrófica crónica y metaplasia intestinal que se han establecido como lesiones precancerosas, debido principalmente al hallazgo de estas características histopatológicas en pacientes con cáncer gástrico(28). En la gastritis atrófica crónica, se observa una reducción o ausencia de las glándulas gástricas normales, un grado variable de inflamación y en ocasiones se observa metaplasia intestinal. La metaplasia intestinal es el reemplazo de las células gástricas normales por células que semejan el epitelio intestinal. A menudo, estas dos entidades coexisten, pero existen casos de metaplasma intestinal aislada sin más hallazgos patológicos. Se conocen dos tipos de metaplasia, el tipo I o tipo completo, se asemeja a la mucosa del intestino delgado con un ribete en cepillo que contiene todas las enzimas habituales presentes en el epitelio intestinal, pero carece de vellosidades. El tipo II o incompleta se subdivide en 2 grupos. El tipo IIa conocida como metaplasia de células caliciales, y el tipo IIb, en que se aprecia un mayor grado de diferenciación celular y que es la que se ha asociado en mayor grado con el cáncer gástrico(28).
La infección por H pylori es una condición que predispone al linfoma MALT. La erradicación de H pylori produce una remisión completa en el 80% de los linfomas de bajo grado (29).
La erradicación de H pylori del aparato gastrointestinal disminuye considerablemente las recidivas de úlceras gastroduodenales. La prevalencia de infección por H pylori es más de 2 veces superior en hijos de madres con antecedentes de úlceras pépticas o duodenales que en madres sanas. La transmisión está completamente justificada por la frecuencia de informes sobre una mayor prevalencia de la infección en hijos de padres infectados(25).
La infestación por la bacteria suele ocurrir durante la infancia y su cuadro clínico se caracteriza por dolor abdominal, náuseas, vómitos mucosos y malestar general.
El cuadro clínico puede extenderse una semana, después de la cual, la sintomatología desaparece permanentemente. Esta enfermedad infecciosa, como muchas otras, puede ser asintomática hasta en el 50% de los adultos. Una vez que la bacteria coloniza el aparato gastrointestinal humano puede producir en pocas semanas o meses una gastritis superficial crónica, que puede culminar en la aparición de una úlcera duodenal, metaplasia intestinal o adenocarcinoma gástrico(30).
Se han descrito además otras patologías extradigestivas en pacientes con infección por H pylori (ver tabla 1). Una de ellas es la anemia ferropénica. Se han reportado niveles de ferritina sérica reducidos en personas con títulos elevados de anticuerpos anti-H pylori. La curación de la infección por H pylori se encuentra asociada con la regresión de la dependencia del hierro y la recuperación de la anemia ferropénica. El tratamiento para la erradicación de la infección por H pylori mejora la anemia aun en pacientes que no reciben terapia con hierro(31).

La infección por H pylori se ha asociado a un mayor riesgo de padecer de enfermedades cardiovasculares: la asociación es independiente de otros factores como hábito de fumar, hipertensión arterial e hiperlipidemias. Una de las hipótesis se centra en la modificación del metabolismo lipídico con aumento de triglicéridos y una reducción de Hdl-colesterol(32). La infección crónica por H pylori, acompañada de inflamación persistente de la mucosa gástrica, incrementa la concentración de proteínas de fase aguda como fibrinógeno y ácido siálico los cuales son predictores de la enfermedad coronaria. Los niveles elevados de homocisteína que se han encontrado en pacientes infestados por H pylori también se han relacionado con incremento del riesgo de arteriosclerosis prematura. La homocisteína inhibe la secreción de óxido nítrico por las células endoteliales lo que facilita la agregación plaquetaria y la vasoconstricción, además puede alterar el balance entre agentes favorecedores e inhibidores de la coagulación sanguínea. La elevación de homocisteína en sangre está relacionada con deficiencia de vitamina B6, B12 y ácido fólico que son necesarias para diferentes pasos de remetilación y transulfuración de la homocisteína(33,34,35,36). Los pacientes con H pylori tienen absorción disminuida de ácido fólico y cobalamina lo que puede incluso generar manifestaciones polineuropáticas sobre todo en aquellos pacientes que presentan concomitantemente infestación por Giardia intestinalis, lo cual predispondría a la acumulación de homocisteína(37).
La infección crónica por H pylori además de que puede precipitar una arteriosclerosis prematura por las alteraciones lipídicas y aumento de homocisteína, puede además agravar el desarrollo de placas de ateromas ya existentes aumentando la activación de las células T, como una respuesta inflamatoria adicional que puede participar en la desestabilización de la íntima de las arterias resultando en la ruptura de la placa y la precipitación de los cuadros isquémicos agudos y el resultante engrosamiento de la íntima(36).
DIAGNÓSTICO DE HELICOBACTER PYLORI
La infección por H pylori puede ser diagnosticada por métodos no invasivos y métodos invasivos y la forma de detectar el microorganismo puede ser directa o indirecta, (ver tabla 2).
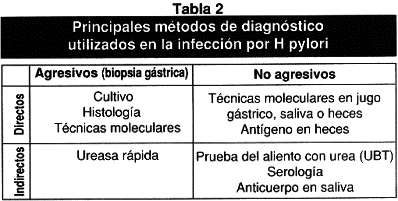
La identificación mediante pruebas histológicas y cultivo provenientes de una muestra de la mucosa gástrica obtenida por endoscopia y la realización de la prueba de ureasa rápida, la reacción de la polimerasa en cadena (PCR) y la tipificación molecular (PCR-RFLP), tienen el inconveniente de la invasividad y por tanto no son aplicables a portadores sanos; adicionalmente representan el resultado local de la muestra del estómago utilizada y no de todo el órgano, por tanto es posible obtener falsos negativos(38). La prueba de urea rápida o CLOtest determina sobre un portaobjeto la presencia de la ureasa producida por H pylori con un simple cambio de color(39,40).
Las proteínas bacterianas inducibles por estrés térmico tienen reacción cruzada con algunos antígenos de los tejidos humanos creando las bases de la autoinmunidad; estas macromoléculas están relacionadas también con los procesos inflamatorios producidos por el microorganismo y esta es el principio de la prueba de ELISA, basado en las proteínas de alto peso molecular asociadas a H pylori. Esta prueba tiene ventajas de costo en estudios epidemiológicos: su intervalo de sensibilidad oscila entre 63-97%. Los estudios serológicos, sin embargo, a pesar de su comprobada eficacia en estudios de terreno presentan el inconveniente de que los anticuerpos una vez que se han producido pueden mantenerse elevados hasta 6 meses después de su erradicación, lo que limita la utilidad de la prueba en los controles de tratamiento(40,41).
H pylori se encuentra también localizado en la boca. Las placas dentarías actúan como reservorio. Estudios de cuantificación están actualmente en curso.
El método considerado en la actualidad como el estándar de oro es la prueba del aliento, que utiliza urea marcada con carbono 13 o carbono 14. La prueba desarrollada por Graham y Klein en 1987 documenta la presencia de la infección momentánea y tiene una respuesta rápida a los efectos de tratamiento y a las reinfecciones que suelen producirse. El paciente ingiere una cápsula que contiene urea y un trazador radioactivo que al hacer contacto con la bacteria es inmediatamente quebrado en dióxido de carbono C14 y amonio ya que H pylori produce ureasa, una enzima ausente en el aparato digestivo alto. El dióxido de carbono entra en el torrente sanguíneo y es filtrado por los pulmones donde es exhalado por el paciente. La muestra de aliento es colectada en un globo. A diferencia de otros métodos, un resultado positivo con la prueba del aliento es confirmatorio de contaminación. Una modificación de este método ha sido propuesta por el Laboratorio de Radioisótopos de la Universidad de Buenos Aires, en el cual se suministra conjuntamente una solución de urea marcada con 14C y un coloide de 99mTc que no se absorbe en el aparato digestivo. Este coloide permite la visualización de la solución de urea dentro del aparato digestivo mediante la utilización de una cámara gamma, lo cual permite localizar exactamente el sitio donde se está produciendo el 14CO2 como consecuencia de la hidrólisis de la urea por el H pylori. Esta combinación de la prueba del aliento con 14C-Urea y la visualización del desplazamiento intragástrico de la solución de Urea-14C permitió elevar la sensibilidad del método al 98% y la especificidad al 96%(42).
TRATAMIENTO DE LA INFECCIÓN POR H PYLORI
El objetivo del tratamiento es la completa eliminación del organismo. Las ratas de reinfección son bajas por lo cual el beneficio del tratamiento es duradero.
La terapia para la erradicación del H pylori debe considerar no solo el uso de antimicrobianos ya que la acidez gástrica influye en la efectividad de algunos antibióticos con actividad contra el H pylori, por lo cual estos deben combinarse con inhibidores de la bomba de protones. En la actualidad el tratamiento de elección para la infección por H pylori es la terapia triple que contenga un inhibidor de la bomba de protones y dos antibióticos(43), (ver Tabla 3).

En 1990 la triple terapia con bismuto fue recomendada en la Conferencia mundial de Gastroenterología en Sidney. Debido a la alta incidencia de efectos secundarios este régimen fue reemplazado por otras triples terapias cortas más eficientes y mejor toleradas(44,45,46).
Estos nuevos regímenes incluyen combinaciones de omeprazol con claritromicina y metronidazol o con claritromicina y amoxicilina, con los cuales se ha reportado una tasa de erradicación del 90% en el estudio MACH-1. Este porcentaje de erradicación ha venido disminuyendo debido a la aparición de cepas de H pylori resistentes al metronidazol que ha llegado a cifras de más del 70% en algunos países como China(47,48), lo cual explica el fracaso de la triple terapia con metronidazol.
En cuanto a la claritromicina, la resistencia reportada ha sido muy variada en el mundo, lo cual está asociado, al amplio uso de este antibiótico en el tratamiento de infecciones respiratorias en algunas ciudades.
En general, se ha observado relativamente alta rata de erradicación con el esquema de claritromicina, amoxicilina y omeprazol y con claritromicina, furazolidona y omeprazol, (90.3% y 90.9% respectivamente). La furazolidona a una dosis de 100 mg dos veces al día(45).
La duración de la terapia permanece en controversia. En Europa, se recomiendan 7 días de tratamiento mientras que en Estados Unidos la triple terapia se extiende por 14 días y es lo aprobado por la FDA. En un meta-ánalisis reciente, se observó que el esquema de tratamiento de 14 días produce una erradicación de H pylori un 8% superior al observado con el régimen de 7 días(49).
La erradicación es más difícil cuando fracasa el primer esquema terapéutico frecuentemente debido a mal cumplimiento del tratamiento por parte del paciente o a la aparición de resistencia. Lo ideal en estos casos sería realizar un cultivo de H pylori con test de susceptibilidad a los antibióticos lo cual es difícil de realizar en la práctica clínica común. Lo recomendado, al no contar con la susceptibilidad antibiótica es prescribir un segundo curso de tratamiento con un inhibidor de la bomba de protones de última generación y rotar los antibióticos, si se usó amoxicilina y claritromicina como primera línea, entonces indicar metronidazol y tetraciclina o viceversa. Podría indicarse adicionalmente a la triple terapia bismuto si no se usó previamente.
El tratamiento de la infección por H pylori en niños tiene algunas particularidades. En general se recomienda la pauta de amoxicilina, metronidazol y bismuto en niños menores de 12 años y la terapia con amoxicilina, claritromicina y omeprazol en mayores de 12 años, dado que la resistencia a la claritromicina es menor a partir de esta edad. Los mejores resultados se han obtenido con pautas de 2 semanas.
COMO ABORDAR LA INFECCIÓN POR H PYLORI?
En el caso de H pylori, la combinación de su relativa frecuencia, la posibilidad de que de lugar a problemas graves y el desconocimiento de cuales son los factores que influyen en su evolución, traen como resultado un alto grado de incertidumbre a la hora de recomendar una actitud concreta en la práctica clínica. Las recomendaciones más recientes son las aportadas por el panel europeo de Maastricht 2-2.000. De acuerdo a este consenso, deben tratarse todos los pacientes con H pylori que tengan úlcera gástrica o duodenal, gastritis atrófica, linfoma MALT, reciente resección de cáncer gástrico, antecedentes familiares de cáncer gástrico. El tratamiento en los pacientes con dispepsia no ulcerosa, enfermedad de reflujo gastroesofágico y pacientes que consumen AINES es opcional, (ver Tabla 4).

En base a el mayor riesgo de cáncer gástrico que tienen los pacientes con infección por H pylori se podría considerar elemento de peso para tratar a todos los pacientes en los cuales se detecte la presencia de H pylori y buscar su completa erradicación.
Aunque se han logrado enormes avances en el estudio de los factores de virulencia de H pylori y sus variaciones genéticas, esta información no ha sido totalmente aplicada a la práctica clínica. Asociaciones entre las características de la bacteria y el riesgo de enfermedad no han permitido definir suficientemente bien una guía de decisiones terapéuticas. La vacunación como profilaxia ha sido exitosa en modelos animales pero su aplicación en humanos ha sido difícil en parte debido a que la inmunología del estómago no es bien conocida. Es necesario seguir profundizando en los estudios.
REFERENCIAS BIBLIOGRÁFICAS
1. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis.Lancet 1983; 1: 1273-1275. [ Links ]
2. Malaty HM, El-Kasabany A, Graham DY, Millar Ch C, Reddy SG, Srinivasan SR, Yasmaoka Y, Berenson gs. Age at acquisition of Helicobacter pylori infection: a follow-up study from infante to adulthood. Lancet 2002; 359: 931-935. [ Links ]
3. Sherman P, Czimm S, Drumm B, Coltrand F, Kawakami E, Machazo A, Oderda G, Seo J-K, Sulliva P, et al. Helicobacter infection in children and adolescents: Working Group Report of the First World Congress of Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2002; 35: S128-S133. [ Links ]
4. Shashidar H, Peters J, Lin Ch, Rabah R, Thomas R, Tolia V. A prospective trial for Pediatric Helicobacter pylori infection. J Ped Gastroenterol Nutr 2000; 30: 276-282. [ Links ]
5. Goodman KJ, Correa P, Tengana Aux HJ, et al. Helicobacter pylori infection in the Colombian Andes: a population-based study of transmission pathways. Am J Epidemiol 1996; 144: 290-5. [ Links ]
6. Dore MP, Sepúlveda AR, El-Zimaity H, et al. Isolation of Helicobacter pylori from sheep-implications for transmission to humans. Am J Gastroenterol 2001; 96: 1396-401. [ Links ]
7. Brenciaglia, et al. Helicobacter pylori: cultivability and antibiotic susceptibility of cocoide forms. IJAA 2000; 13: 237-241. [ Links ]
8. Petersen AM, Krogfelt KA. Helicobacter pylori: an invading microorganism? A review. Inmunol Med Microbiol 2003; 36(3): 117-26. [ Links ]
9. Olczak AA, et al. Association of Helicobacter pylori antioxidant activities with host colonization proficiency. Infect Immun 2003; 71: 580-583. [ Links ]
10. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 1273-5. [ Links ]
11. Tummuru MKR, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun 1993; 61: 1799-809. [ Links ]
12. Mai UEH, Perez-Perez GI, Allen JB, Wahl SM, Blaser MJ. Surface proteins from Helicobacter pylori exhibit chemotactic activity for human leukocytes and are present in gastric mucosa. J Exp Med 1992; 175: 517-25. [ Links ]
13. Heikkinen M, Mayo K, Megraud F, Vornanen M, Marin S, Pikkarainen P, Julkunen R. Association of Cag-A positive and Cag-A negative Helicobacter pylori strains with patients symptoms and gastritis in primary care patients with functional upper abdominal complaints. Scan J Gastroenterol 1998; 33: 31-38. [ Links ]
14. Zhu YL, Zheng S, Qian KD, Fang PC. Biological activity of the virulence factor CagA of Helicobacter pylori. Chin Med J (Engl) 2004; 117(9): 1330-3. [ Links ]
15. Zulio A, sanchez-Mete L, Hassan C, Diana F, Festuccia F, Attili AF, Morini S. Helicobacter pylori density and CagA status in cirrhotic patients: A case-control study. J Gastroenterol Hepatol 2004; 19(10): 1174-1178. [ Links ]
16. Hofman P, Waidner B, Hofman V, Bereswill S, Brest P, Kist M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2004; 9(suppl 1): 15-22. [ Links ]
17. Blaser MJ. The genetic Gymnastics of Our Indigenous Microbes. N Eng J Med 2002; 346: 2083-85. [ Links ]
18. Negrini R, Savio A, Appelmelk BJ. Autoantibodies to gastric mucosa in Helicobacter pylori infection. Helicobacter 1997; 2(Suppl 1): S13-S16. [ Links ]
19. Lin HJ, Perng CL, Lo WC, Wu CW, Tseng GY, Li AF, Sun IC, Ou YH. Helicobacter pylori cagA, iceA and vacA genotypes in patients with gastric cancer in Taiwan. World J Gastroenterol 2004; 10(17): 2493-7. [ Links ]
20. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4(9): 688-94. [ Links ]
21. Zhang QB, Nakashabendi IM, Mokhashi MS, Dawodu JB, Gemmell CG, Russel RI. Association of cytotoxin production and neutrophil activation by strains of Helicobacter pylori isolated from patients with peptic ulceration and chronic gastritis. Gut 1996; 38: 841-5. [ Links ]
22. Rudi J, Kuck D, Strand S, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest 1998; 102: 1506-14. [ Links ]
23. El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2001; 412-99. [ Links ]
24. S Lorente, O Doiz, M Trinidad Serrano, J Castillo and A Lanas. Helicobacter pylori stimulates pepsinogen secretion from isolated human peptic cells. Gut 2002; 50: 13-18. [ Links ]
25. Martin JB. The role of H. pylori in gastritis and its progress to peptic ulcer disease. Aliment Pharmacol Ther 1995; 9: 27-30. [ Links ]
26. Jaakimainen RL, Boyle E, Tudiver F. Is Helicobacter pylori associated with non-ulcer dyspepsia and will eradication improve symptoms? A meta-analysis. BMJ 1999; 319: 1040-4. [ Links ]
27. Uemura N, Okamoto S, Yamamoto S. Helicobacter pylori infection and the development of gastric cancer.N Engl J Med 2001; 345: 784-789. [ Links ]
28. MM Walker. Is intestinal metaplasia of the stomach reversible? Gut 2003; 52: 1-4. [ Links ]
29. M Stolte, E Bayerdörffer, A Morgner, B Alpen, T Wündisch, C Thiede and A Neubauer.Helicobacter and gastric MALT lymphoma. Gut 2002; 50: 19-24. [ Links ]
30. Dixon ME. Pathology of gastritis and peptic ulceration. In: Mobley HLT, Mendz GL, Hazell SL, eds. Helicobacter pylori: physiology and genetics. Washington, D.C: ASM Press, 2001: 459-469. [ Links ]
31. Howden CW, Leontiadis GI. Extragastric manifestations of Helicobacter pylori are they relevant? In: Hunt RH, Titgat GN, eds. Helicobacter pylori: basic mechanisms to clinical cure 2.000. Dordrecht, the Netherlands: Kluwer Academic, 2000: 315-26. [ Links ]
32. Laurila A, Bloigu A, Nayha S, Hassi J, Leinonen M, Saikku P. Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis 1999; 142: 207-10. [ Links ]
33. Markle HV. Coronary artery disease associated with Helicobacter pylori infection is at least partially due to inadequate folate status. Med Hypotheses 1997; 49: 289-92. [ Links ]
34. Muhlestein JB. Bacterial infections and atherosclerosis. J investig Med 1998; 46: 396-402. [ Links ]
35. Tiran A. Helicobacter pylori and citomegalovirus infection and atherosclerosis: An assesment of the current status. Wien Med Wochen 2001; 151: 587-589. [ Links ]
36. Stamler JS, Osborne JA, Jaraki M, Rabbini LE, Mullins M, Singel D, et al. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest 1993; 91: 308-18. [ Links ]
37. Brieva L, Ara JR, Bertol V, Canellas A, Agua C. Polyneuropathy caused by vitamin B12 deficiency secondary to chronic atrophic gastritis and giardiasis. Rev Neurol 1998; 154: 1019-20. [ Links ]
38. Logan RPH, Walker MM. Epidemiology and diagnosis of Helicobacter pylori infection. BMJ 2001; 323: 920-922. [ Links ]
39. Pérez-Pérez GI, Brown WR, Cover TL, Dunn BE, Cao P, Martin JB. Correlation between serological and mucosal inflammatory responses to H. pylori. Clin Diagn Lab Immunol 1994; 1: 325-9. [ Links ]
40. Oderda G,Rapa A, Boldorini R, Bozzola C, Zavallone A, Stringini L, Scherbakova MY, Scherbakov P. Non-invasive test to diagnose Helicobacter pylori infection in very young Children: Gut 2001; 39: A75. [ Links ]
41. Alvarez L, Mendoza M, Marquez L, Rojas E. Infección por Helicobacter pylori en niños que acuden a la emergencia del Hospital José Gregorio Hernandez de Trujillo, Venezuela. Rev Soc Ven Microbiol 2003; 23: 7-8. [ Links ]
42. Zubillaga M, Oliverti P, Calcagno ML, Coldman C, Caro R, Mitta A, et al. Min 14C-UBT: A combination of gastric basal transit and 14C-Urea Breath Test for the detection of Helicobacter pylori infection in human beings. Nuclear Med Biol 1997; 24: 565-9. [ Links ]
43. Gisbert JP, Pajares JM. Helicobacter pylori therapy: first-line options and rescue regimen. Dig Dis 2001; 19: 134-43. [ Links ]
44. Qasim A, O Morain CA. Review article: treatment of Helicobacter pylori infection and factors influencing eradication. Aliment Pharmacol Ther 2002; 16: 24-30. [ Links ]
45. Chuan-Yong Guo, Yun-Bin Wu, Heng-Lu Liu, Jian-Ye Wu, Min-Zhang Zhong. Clinical evaluation of four one-week triple therapy regimens in eradicating Helicobacter pylori infection. World J Gastroenterol 2004; 10(5): 747-49. [ Links ]
46. Engin Altintas, Orhan Sezgin, Oguz Ulu, Ozlem Aydin, Handan Camdeviren. Maastricht II treatment scheme and efficacy of different proton pump inhibitors in eradicating Helicobacter pylori. World J Gastroenterol 2004; 10(11): 1656-1658. [ Links ]
47. Jenks PJ, Edwards DI. Metronidazole resistancie in Helicobacter pylori. In J Antimicrob Agents 2002; 19: 1-7. [ Links ]
48. Unge P. The OAC and OMC options. Eur J Gastroenterol Hepatol 1999; 11: 23-24. [ Links ]
49. Calvet X, García N, Lopez T, Gisbert JP, Gene E, Roque M. A meta-analysis of short versus long therapy with a proton pump inhibidor, clarithromicin and either metronidazole or amoxycilin for treating Helicobacter pylori infection. Aliment Pharmacol Ther 2000; 14: 603-9. [ Links ]
