Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO  uBio
uBio 
 Permalink
PermalinkRevista Científica
versão impressa ISSN 0798-2259
Rev. cient. (Maracaibo) v.20 n.4 Maracaibo jul. 2010
EXTRACCIÓN DE OXITETRACICLINA EN CARNE DE POLLO: ESTUDIOS DE RENDIMIENTO CON AUMENTO DE LA FASE POLAR DEL SOLVENTE DE EXTRACCIÓN
Pedro Izquierdo 1, Ronald Mavárez 2, Fredy Ysambertt 2, María Ysabel Piñero 1, Gabriel Torres 1 y María Allara 1
1 Unidad de Investigación Ciencia y Tecnología de Alimentos (UDICTA), Facultad de Ciencias Veterinarias.
2 Facultad Experimental de Ciencias. Universidad del Zulia. marysa_pg@yahoo.com; allara2004@hotmail.com
RESUMEN
El consumo de alimentos con residuos de oxitetraciclina (OTC) puede causar diversos efectos tóxicos en el humano. Con la finalidad de extraer y cuantificar dichos residuos en matrices biológicas, como carne de pollo, se han desarrollado diversos métodos. Dentro de los métodos propuestos el más empleado es la extracción líquido-líquido por ser sencillo, rápido y económico. Este tipo de extracción fue aplicada por Furusawa para OTC en pollo, empleando acetonitrilo/hexano en una proporción 5:4 obteniendo una recuperación del 88%. Este trabajo tuvo como objetivo estudiar la recuperación de OTC en carne de pollo ensayada por Furusawa, aumentando la proporción del solvente polar con respecto al hexano (2:1), para su posterior cuantificación mediante cromatografía líquida de alta resolución (HPLC). Para ello se utilizaron 24 porciones de 1 g de tejido muscular perteneciente al muslo de 3 pollos libres de antibióticos, las cuales se fortificaron con soluciones estándares de OTC de 0,1; 0,2; 0,5 y 1 µg/g, obteniendo 6 muestras fortificadas con cada concentración, las cuales fueron almacenadas por 12 horas a 4°C. La extracción del antibiótico se llevó a cabo con acetonitrilo/hexano en proporciones 5:4 y 2:1. En cada caso se evaluó la recuperación, precisión y sensibilidad. Tanto para la proporción 5:4 como 2:1, la concentración de 0,2 µg/g presentó la mayor recuperación, siendo 91,5 y 92,5%, respectivamente; sin embargo, al aumentar la concentración de OTC disminuyó la recuperación. La precisión se incrementó a la concentración de 0,5 µg/g, sin embargo, al duplicar la concentración a 1 µg/g disminuyó dicho parámetro. El límite de detección obtenido para la extracción de OTC con acetonitrilo/hexano en proporción 2:1 fue de 0,09 µg/g. Se recomienda realizar una desproteinización de la muestra previo al proceso de extracción.
Palabras clave: Oxitetraciclina, pollo, rendimiento, HPLC.
Oxytetracycline Extraction in Chicken Meat: Studies on Yield increasing Polar
Phase in Extraction Solvent
ABSTRACT
Consumption of food that contains oxytetracycline (OTC) residues may produce several toxic effects in human beings. In order to extract and quantify such residues in biological matrices, like chicken meat, several methods have been developed. Among these methods, liquid-liquid extraction is the mostly used, because is quick, simple and inexpensive. This extraction method was applied by Furusawa for OTC in chicken, using acetonitrile/hexanes in a 5:4 proportion, obtaining an 88% recovery. The objective of this research was to study extraction of OTC in chicken meat assayed by Furusawa, raising polar solvent proportion in relation to hexanes (2:1), and further quantifying by means of high performance liquid chromatography (HPLC). Twenty four portions of 1 g from 3 OTC free chickens were fortified with OTC standard solutions: 0.1, 0.2, 0.5 and 1 µg/g, obtaining 6 fortified samples for each concentration, stored for 12 hours at 4°C. Antibiotic extraction was performed using acetonitrile/hexanes in 5:4 and 2:1 proportions. Recovery, precision and sensitivity were analyzed in all samples. Either 5:4 or 2:1 proportions, 0.2 µg/g concentration obtained the higher recovery, 91.5 and 92.5%, respectively; however when OTC concentrations raised, recovery became lower. Precision increased at 0.5 µg/g concentration, but, fell down when concentration duplicated: 1 µg/mL. Detection limit obtained for OTC extraction using acetonitrile/hexanes in 2:1 proportion were 0.09 µg/g. Deproteinization is recommended previously to extraction process.
Key words: Oxytetracycline, chicken, yield, HPLC.
Recibido: 03 / 12 / 2008. Aceptado: 17 / 03 / 2010.
INTRODUCCIÓN
La oxitetraciclina (OTC) es un antibiótico perteneciente al grupo de las tetraciclinas de primera generación, que deriva del anillo policíclico naftalenocarboxamida. Este compuesto se caracteriza por ser bacteriostático, aunque puede llegar a ser bactericida cuando se administra en altas concentraciones. Una vez que el antibiótico ingresa a la bacteria por difusión pasiva y transporte activo, inhibe la síntesis proteica evitando la reproducción del microorganismo [17, 20, 26].
La OTC se aplica en la medicina veterinaria para el tratamiento y prevención de enfermedades infecciosas que afectan la población animal. Además, se emplea como promotor del crecimiento, incrementando la productividad y el consecuente beneficio económico para la industria avícola [3, 14]. Sin embargo, la administración de OTC a un animal, puede provocar que sus residuos estén presentes en alimentos derivados de éstos, como carne, leche, huevo, entre otros, los cuales al ser consumidos frecuentemente por personas susceptibles pueden causar efectos tóxicos como reacciones alérgicas cutáneas, náuseas, vómitos, choque anafiláctico e incluso la muerte. Además, su uso indiscriminado puede generar resistencia bacteriana al antibiótico [6, 22, 23, 27].
Los antibióticos presentan diversas propiedades que los diferencian químicamente entre sí, por lo tanto se requiere el uso de métodos de análisis específicos en cada caso. Tradicionalmente se han empleado ensayos microbiológicos, sin embargo, estos métodos presentan dificultades en la selectividad y especificidad para el análisis de antibióticos de una misma familia [13, 16]. En tal sentido, los métodos de análisis cromatográfico ofrecen una respuesta rápida, de alta sensibilidad y una separación eficiente de todos los análogos de una familia o grupo de antibióticos, lo cual permite que cada especie interaccione de manera particular con la fase móvil y estacionaria del sistema cromatográfico [11, 24].
Diversos métodos han sido desarrollados y validados para la extracción y cuantificación de OTC en diferentes matrices biológicas [1, 4, 8-10, 13, 16, 18, 21, 28]. Los métodos de extracción y purificación utilizan técnicas como: quelación de las tetraciclinas con iones de metales divalentes [8], extracción en fase sólida [1, 18, 28], extracción líquido-líquido [9], entre otros. La extracción líquido-líquido es la más aplicada para la determinación de tetraciclinas, debido a que es un procedimiento sencillo, rápido y económico, sin embargo, existe la necesidad de mejorar su rendimiento, ya que el porcentaje de recuperación suele ser inferior al 90% [9].
Entre los métodos propuestos para la extracción y cuantificación de OTC en pollo por cromatografía líquida de alta resolución (HPLC) que emplean extracción líquido-líquido se encuentra el método propuesto por Furusawa, el cual permite la determinación de este antibiótico con un porcentaje de recuperación del 88% [9]. El objetivo de este trabajo fue optimizar la recuperación del proceso de extracción de la OTC en carne de pollo ensayada por este autor, aumentando la proporción del solvente polar con respecto al hexano y su posterior cuantificación por cromatografía líquida.
MATERIALES Y MÉTODOS
Preparación de estándares
Se preparó una solución madre de OTC (marca Sigma®) en acetonitrilo a una concentración de 100 µg/mL, y luego por dilución se obtuvo una solución intermedia de 10 µg/mL. A partir de la solución intermedia se prepararon soluciones de trabajo con concentraciones de 0,1; 0,2; 0,5; 0,8 y 1,0 µg/mL.
Preparación de las muestras
Las muestras se obtuvieron a partir de 3 pollos, los cuales fueron criados sin recibir tratamiento antimicrobiano (incluyendo OTC) y posteriormente sacrificados a las 6 semanas de vida. Para el análisis se utilizó el tejido muscular proveniente del muslo. Se pesaron 24 porciones de 1 g de cada pollo, las cuales se enriquecieron con soluciones estándares de OTC hasta obtener concentraciones de 0,1; 0,2; 0,5 y 1,0 µg/g, 6 muestras para cada concentración. Las muestras fortificadas fueron almacenadas por 12 horas a 4°C [28] en un refrigerador, marca Tropicold, modelo VS4P, fabricado por Neve Industrial C.A (Venezuela).
Extracción de la OTC
Las muestras fueron homogenizadas con 25 mL de acetonitrilo saturado con n-hexano y 20 mL de n-hexano en un homogenizador, marca Ultra-Turrax T8®, modelo S8N-5G (Alemania), a máxima velocidad durante 2 min. El extracto se centrifugó a 1.500 g por 5 min en una centrífuga, marca Dynac®, modelo 0101 (EUA) [9]. El sobrenadante se aisló por filtración con papel filtro Whatman N° 1 y se reservó la capa polar (acetonitrilo) en un embudo de separación. Ésta fue colectada en un balón de destilación de 100 mL y evaporada a sequedad en un baño María, modelo YCW-01, fabricado por Gemmy Industrial Corp. (Taiwán) a 45°C, empleando un rotoevaporador, marca Rinco®, modelo VE1002B (EUA). El residuo fue disuelto en 1 mL de acetonitrilo. La solución se filtró a través de una membrana Millipore® de 0,22 µm de poro [9]. Dicho procedimiento se realizó de igual manera utilizando durante la homogenización 40 y 20 mL de acetonitrilo y n-hexano, respectivamente, para así obtener una proporción de 2:1.
Condiciones cromatográficas
Se empleó un equipo de HPLC de la serie 6A marca Shimadzu® (Japón). El inyector utilizado fue marca Rheodyne®, modelo 7125 (EUA), equipado con un loop de 20 µL. La fase móvil estuvo constituida por acetonitrilo: ácido acético al 5% (35:65) [9], en un sistema isocrático. La misma fue filtrada a través de una membrana Millipore® de 0,45 µm de poro y desgasificada por 10 min con burbujeo de nitrógeno y sonicación al vacío. Se utilizó una columna de fase reversa, marca Merck Lichrospher RP8 con dimensiones de 250 mm x 4,5 mm ø x 5 µm (Alemania). La temperatura de la columna fue 26°C y el flujo fue 1,0 mL/min. Se empleó un detector ultravioleta SPD 10-A, marca Shimadzu® (Japón), a una longitud de onda de 357 nm [9]. El procesamiento de los datos se realizó con el software cromatográfico Class VP, marca Shimadzu, versión 4.1, suministrado por la misma casa comercial.
Determinación de parámetros analíticos
Para determinar la linealidad del método se construyó una curva de calibración a través del método de mínimos cuadrados, donde se relacionó la concentración del analito con el área del pico. La precisión del método se determinó evaluando los siguientes parámetros: desviación estándar (s), varianza (s2), desviación estándar relativa (DER) y coeficiente de variación (CV).
La sensibilidad analítica fue determinada considerando la expresión g= m/s [15, 24]. Donde: m es la pendiente de la regresión por método de mínimos cuadrados y s es la desviación estándar de las medidas. El límite de detección (LD) se determinó como la concentración de analito que proporciona una señal igual a la señal del blanco (yB), más tres veces la desviación estándar del blanco (SB), lo cual se describe en la siguiente ecuación: y=3SB+ yB. El límite de cuantificación (LC) fue calculado de manera similar al LD, pero multiplicando el valor de desviación estándar por un factor igual a 10 [15, 19].
RESULTADOS Y DISCUSIÓN
Extracción de la OTC
La extracción del antibiótico se llevó a cabo usando la combinación de los solventes acetonitrilo y hexano en proporciones 5:4 y 2:1. En ambos casos fue necesario realizar un proceso de filtración por gravedad previo a la separación de los líquidos inmiscibles, debido a que algunas partículas de la matriz eran arrastradas hacia el embudo de separación dificultando el proceso de extracción del analito. Por otra parte, se ensayaron varias temperaturas (45; 50 y 55°C) obteniéndose una pérdida de la respuesta proporcional al aumento de ésta, lo cual sugiere que ocurrió una descomposición o cambio estructural del antibiótico durante el secado. La temperatura máxima de secado, que no afectó la respuesta del método fue de 45°C, por lo tanto, todas las muestras fueron secadas a esta temperatura en baño María. Tanto el proceso de filtración, como la temperatura de secado fueron establecidas en este estudio, debido a que en la metodología original propuesta por Furusawa [9] no se detallan los valores de temperatura utilizados para concentrar el extracto, ni se especifican procedimientos para preparar la muestra previa a una ultrafiltración.
Durante la etapa de resuspensión del analito en la fase móvil se observó la formación de un precipitado de aspecto coloidal. Para determinar la naturaleza del precipitado se realizó el ensayo de Bradford [2], el cual resultó positivo a la presencia de proteínas. Esta precipitación pudiera ser ocasionada por el efecto del ácido acético presente en la fase móvil sobre las proteínas contenidas en el extracto. Aunque Furusawa [9] no describe de manera explícita en su método esa interferencia, hace la salvedad que durante la extracción de OTC en huevo no se presenta ninguna turbidez, debido a que se realiza una filtración seguida de la resuspensión en acetonitrilo, en lugar de emplear fase móvil.
Separación de la OTC
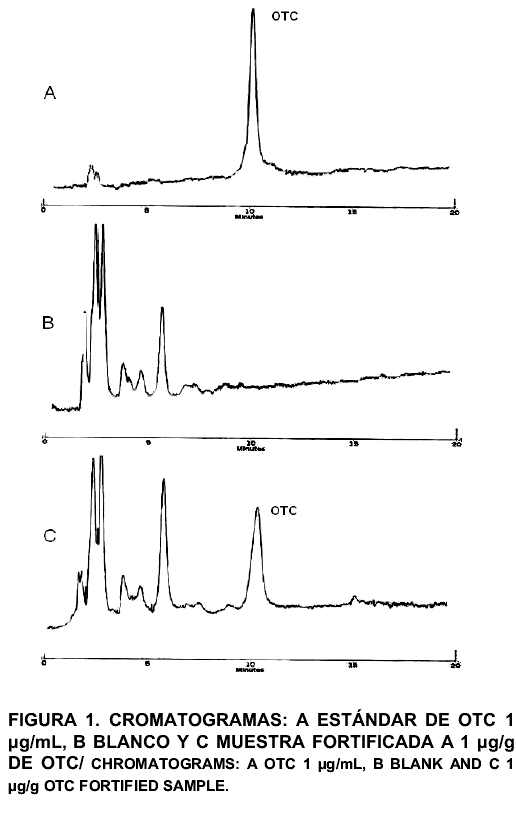
La FIG. 1 muestra la separación de OTC utilizando el método propuesto en este trabajo. El analito presentó un tiempo de retención de 10,1 min como se muestra en el cromatograma A, correspondiente a un estándar de OTC con una concentración de 1 µg/mL, observándose un pico simétrico y definido. El cromatograma B corresponde a un extracto de carne de pollo libre de OTC utilizado como blanco y el C pertenece a un extracto fortificado con 1 µg/g de OTC. Los compuestos que absorben luz UV a la longitud de onda utilizada en el análisis (357 nm), aparecen antes de los 7 min, por lo que no interfieren con la salida del pico de OTC (cromatograma C).
Recuperación
La TABLA I muestra la comparación de las recuperaciones de OTC con acetonitrilo/hexano en proporciones 5:4 y 2:1, a las diferentes concentraciones ensayadas. La recuperación del método de extracción expresada en porcentaje (%R) estuvo entre 91,5 y 31,4% empleando la proporción 5:4, mientras que para la proporción 2:1 osciló entre 92,5 y 64,5%. En ambos casos, la muestra fortificada con 0,2 µg/g de OTC presentó la mayor recuperación (91,5 y 92,5%), sin embargo, al aumentar la concentración del analito se presentó una disminución en la recuperación. Por otro lado, se observó una relación inversa entre el %R y la masa fortificada con el analito a extraer. Dicha pérdida pudiera atribuirse a la absorción del analito en el papel de filtro debido a la alta capacidad de absorción de la celulosa [29]. Otra fuente de absorción del analito es el tamaño de las partículas de cortes en el proceso de homogenización debido al área de superficie por unidad de masa de un sólido (área de superficie específica), expresada generalmente en centímetros cuadrados por gramo (cm2/g). Para la masa de un sólido, el área de superficie específica aumenta drásticamente cuando se reduce el tamaño de la partícula. Por lo tanto, al aumentar el área de superficie aumenta la absorción del analito [25], lo cual indica que existe una pérdida o retención del analito durante la extracción.
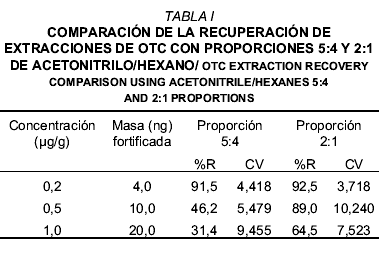
Cabe destacar que, con las proporciones de solventes ensayados, la recuperación del antibiótico fue mayor con respecto a otros métodos de extracción. El empleo de acetonitrilo como solvente en el proceso de extracción de la OTC, obedece a la gran solubilidad del antibiótico en el mismo. La OTC tiene en su estructura un grupo amino y amido, cada uno de los cuales presenta un par de electrones no enlazados, lo que permite afinidad e interacción entre éste y el acetonitrilo, además de las diferentes insaturaciones del anillo naftalénico que pudieran interactuar con la insaturación del acetonitrilo [10].
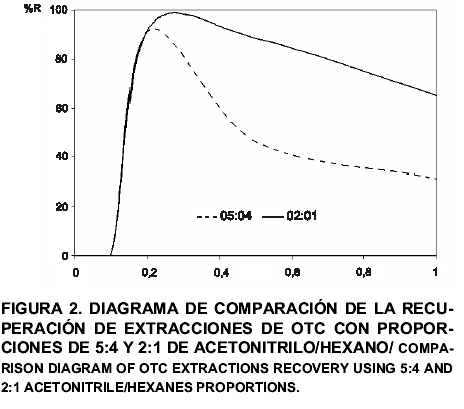
La FIG. 2 muestra un diagrama de comparación de la recuperación del antibiótico a las diferentes concentraciones del mismo. A pesar de que se observa una disminución en la recuperación del antibiótico al aumentar la concentración del mismo, con las proporciones de solventes ensayadas se duplica la recuperación del antibiótico con respecto a la proporción de acetonitrilo: hexano propuesta por Furusawa [10].
Parámetros analíticos del método
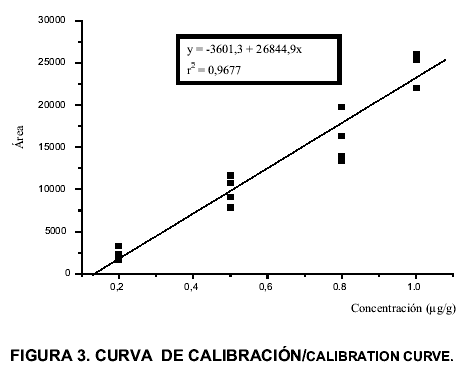
En la FIG. 3 se muestra la curva de calibración obtenida, observándose una respuesta lineal de las áreas en el intervalo de concentraciones entre 0,2 y 1,0 µg/g (r2 = 0,9677). En la TABLA II se observan los parámetros de precisión (s, s2, DER, CV) determinados en este estudio, los cuales cuantifican el grado de concordancia entre los datos obtenidos bajo las mismas condiciones. En la extracción de OTC empleando proporciones de acetonitrilo/hexano de 5:4, se observó un aumento progresivo del grado de dispersión en el análisis, proporcional a la concentración del analito y a la masa extraída, mientras que para las extracciones con proporciones de 2:1, la precisión se incrementó a la concentración de 0,5 µg/g, sin embargo al duplicar la concentración a 1,0 µg/g se observó una disminución de dicho parámetro, es decir, que el método propuesto es mas preciso mientras mas baja sea la concentración a detectar, lo cual resulta conveniente ya que esa concentración está mas cercana al límite máximo de residuos sugerido (LMRs) para OTC en pollo.
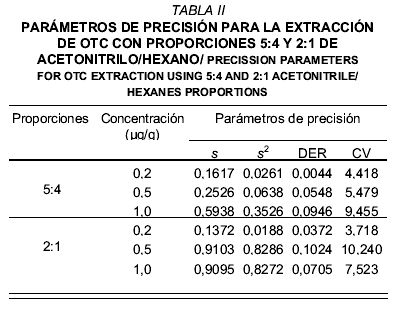
El límite de cuantificación obtenido para el método propuesto en este estudio fue de 0,2 µg/g, mientras que el límite de detección fue de 0,09 µg/g, siendo éste último un valor inferior al establecido como LMRs para OTC en músculo de pollo (100 µg/kg) por diversos organismos oficiales [5, 7, 12].
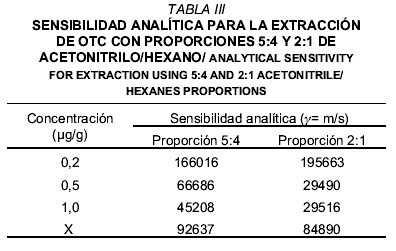
La TABLA III muestra los valores de sensibilidad analítica obtenida con las proporciones de solventes ensayadas a diferentes concentraciones del analito. La proporción de acetonitrilo/hexano 5:4 resultó ser analíticamente más sensible que la proporción 2:1, ya que presenta mayor capacidad de diferenciar pequeñas variaciones en la concentración del analito. Esto se relaciona con los resultados obtenidos para la precisión, ya que para la proporción de solventes 2:1 fue mayor la dispersión de los datos, lo cual es inversamente proporcional a la sensibilidad del método. En tal sentido, diversos autores [19, 24] refieren que la sensibilidad de un método depende de dos factores: la pendiente de la curva de calibración y la precisión del sistema de medida. Aunado a ello, se considera que entre dos métodos que tengan curvas de calibración con igual pendiente, será más sensible aquel que presente la mejor precisión.
CONCLUSIONES Y RECOMENDACIONES
La modificación realizada para la extracción de OTC en carne de pollo es altamente recomendada, dado que permite una mayor recuperación del analito en comparación con el método propuesto por Furusawa.
La técnica desarrollada para la extracción de OTC en carne de pollo representa un método sencillo y rápido, debido a que implica procedimientos de fácil ejecución en un laboratorio de rutina.
El límite de detección obtenido en este estudio (0,09 µg/g), aunque fue superior al reportado por Furusawa (0,05 µg/g), permite detectar concentraciones de OTC inferiores a los LMR sugeridos por organismos oficiales internacionales.
Incluir dentro del proceso de extracción líquido-líquido de OTC la desproteinización previa de la muestra o extracción en fase sólida, para garantizar la vida útil de la columna cromatográfica, eliminar compuestos interferentes del sistema en general y reducir el volumen de solventes empleados.
Aplicar el método de extracción de OTC empleando acetonitrilo/hexano en proporciones de 2:1 y cuantificarlo con un detector de espectroscopia de emisión y/o fluorescencia.
Aplicar el método de extracción para OTC a otras tetraciclinas empleadas en la industria avícola utilizando acetonitrilo/hexano en proporciones de 2:1.
AGRADECIMIENTO
Los autores expresan su agradecimiento al Consejo de Desarrollo Científico y Humanístico de la Universidad del Zulia (CONDES-LUZ) por el financiamiento de esta investigación.
REFERENCIAS BIBLIOGRÁFICAS
1. BLASCO, C.; DI CORCIA, A.; PICÓ, Y. Determination of tetracyclines in multi-specie animal tissues by pressurized liquid extraction and liquid chromatography–tandem mass spectrometry. Food Chem 116 (4) 1005–1012. 2009. [ Links ]
2. BRADFORD, M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 7 (72): 248-254. 1976. [ Links ]
3. CANCHO, B.; GARCÍA, M.; SIMAL, J. El uso de los antibióticos en la alimentación animal: Perspectiva actual. Cien. Tecnol. Aliment. 3 (1): 39-47. 2000. [ Links ]
4. CAPOLONGO, F.; SANTI, A.; TOMASI, L.; ANFOSSI, P.; MISSAGIA, M.; MONTESISSA, C. Residues of oxytetracycline and its 4-epimer in edible tissues from turkeys. J. AOAC Int. 85 (1): 8-14. 2002. [ Links ]
5. COMMISSION REGULATION 508/1999/EC. Amending Annexes I–IV to Council Regulation (EEC) No 2377/90 laying down Community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origen. Off. J Eur. Commun. L224, 16. 1999. [ Links ]
6. DAZA, R. Resistencia bacteriana a antimicrobianos: su importancia en la toma de decisiones en la práctica diara. Inf. Ter. Sist. Nac. Salud 22:57-67. 1998. [ Links ]
7. FAO/WHO FOOD STANDARDS CODEX ALIMENTARIUS. Veterinary drug residues in food. Maximum residue limits. 2006. On line: <http:// www.codexalimentarius.net/mrls/vetdrugs/jsp/vetd_q-e.jsp. 06/10/2006. [ Links ]
8. FARRINGTON, W.; TARBIN, J.; BYGRAVE, J.; SHEARER, G. Analysis of trace residues of tetracyclines in animal tissues and fluids using metal chelate affinity chromatography/HPLC. Food Addit. Contam. 8 (1):55-64. 1991. [ Links ]
9. FURUSAWA, N. Rapid and simple determination of oxytetracycline in chicken products. J. AOAC Int. 82 (3): 770-772. 1999. [ Links ]
10. FURUSAWA, N. Simplified Liquid-Chromatographic determination of residues of tetracycline antibiotics in eggs. J. Chromatogr. 53 (1): 47-50. 2001. [ Links ]
11. HARVEY, D. Métodos cromatográficos y electroforéticos. Química Analítica Moderna. 1ra Ed. Mc Graw Hill, E.U.A. Pp 402-409. 2002.
12. JAPANESE MINISTRY OF HEALTH WELFARE AND LABOR. Food safety information. 2010. On line: ttp://www. mhlw.go.jp/english/topics/foodsafety/index.html. 11/03/2010.
13. JEVINOVA, P.; DUDRIKOVA, E.; SOKOL, J.; NAGY, J.; MATE, D.; PIPOVA, M.; CABADAJ, R. Determination of oxytetracycline residues in milk with the use of HPLC method and two microbial inhibition assays. Bull. Vet. Inst. Pulawy. 47 (1):211-216. 2003. [ Links ]
14. LARA, J.; ECHEVERRIA, P.; CARVAJAL, M. Residuos de antibióticos en carne e hígado de cerdos y aves que se consumen en la ciudad de Mérida. Vet. Méx. 22 (2):53-56. 1991. [ Links ]
15. MILLER, J.C.; MILLER, J.N. Métodos de calibración en análisis instrumental: regresión y y correlación. Estadística y Quimiometría para Química Analítica. 4ta Ed. Prentice-Hall, España. Pp 111-155. 2002.
16. MOATS, W. Determination of tetracycline antibiotics in beef and pork tissues using Ion-Paired Liquid Chromatography. J. Agric. Food Chem. 48 (6): 2244-2248. 2000. [ Links ]
17. MOREJÓN, M.; SALUP, R.; CUE, M. Actualización en tetraciclinas. Rev. Cub Farm. 37 (3): 1-1. 2003. [ Links ]
18. NIKOLAIDOU, KI; SAMANIDOU, VF; PAPADOYANNIS, IN. Development and validation of an HPLC method for the determination of seven tetracycline antibiotics residues in chicken muscle and egg yolk according to 2002/657/EC. J liquid Chromatogr & Relat Technol. 31 (14): 2141-2158. 2008. [ Links ]
19. QUATTROCCHI, O.; ABELARIA, S.; LABA, R. Validación de métodos. Introducción a la HPLC. Aplicación y Práctica. Grafias Farro, Argentina. Pp 302-327. 1992.
20. RANG, H.P.; DALE, M.M. Agentes antimicrobianos que afectan a la síntesis de las proteínas bacterianas. Farmacología. 3ra Ed. Madrid, Eds Churchill Livingstone. Pp 885-892. 1996.
21. SCHNEIDER, M.; BRADEN, S.; REYES-HERRERA, I.; DONOGHUE, D. Simultaneous determination of fluoroquinolones and tetracyclines in chicken muscle using HPLC with fluorescence detection. J. Chromatogr B. 846 (1-2): 8-13. 2007. [ Links ]
22. SHERWOOD, L.; GORBACH, M. Antimicrobial use in animal feed – Time to stop. N Engl. J. Med. 345 (16):1202-1203. 2001. [ Links ]
23. SINGH, M.; VARSHNEYA, C.; TELANG, R.; SRIVASTAVA, A. Alteration of pharmacokinetics of oxytetracycline following oral administration of Piper longum in hens. J. Vet. Sci. 6 (3): 197-200. 2005. [ Links ]
24. SKOOG, D.; HOLLER, F.; NIEMAN, T. Introducción a las separaciones cromatográficas. Principios de Análisis Instrumental. 5ª Ed. McGraw-Hill, España. Pp 785-787. 2000.
25. SKOOG, D.; WEST, D.; HOLLER, J. Titulaciones por formación de complejos. Química Analítica. 6ª Ed., Traductor: Ramírez Carmen. Mexico, McGraw-Hill. Pp 237-252. 1995.
26. SUMANO, H.; OCAMPO, L. Antimicrobianos. Farmacología Veterinaria. 2ª Ed., México, Mc Graw-Hill Internacional. Pp 95-98. 1997.
27. TOLLEFSON, L.; MILLER, M. Antibiotic use in food animals: controlling the human health impact. J. AOAC Int. 83 (2):245-254. 2000. [ Links ]
28. WALSH, J.; WALKER, L.; WEBBER, J. Determination of tetracyclines in bovine and porcine muscle by high-performance liquid chromatography using solid-phase extraction. J. Chrom. 596 (2): 211-216. 1992. [ Links ]
29. YANG, L.; SHIJIE, L. Kinetic model for kraft pulping process. Ind. Eng. Chem. Res. 44 (18):7078-7085. 2005. [ Links ]