










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Investigación
versión impresa ISSN 1010-2914
Revista de Investigación vol.36 no.76 Caracas ago. 2012
Modelaje Molecular de E-1-[4-(3-(sustituido) acriloil) fenil]-3-tosilureas
Molecular Modeling of E-1[4-(3-(substituted) acryloyl) phenyl]-3-tosylureas
Caritza León(1), Trina Colman(2), José Domínguez(3)
(1) Universidad Pedagógica Experimental Libertador. Instituto Pedagógico de Caracas. Caracas, Venezuela. caritzal@cantv.net
(2) Laboratorio de Modelaje Molecular. Universidad Central de Venezuela. Caracas, Venezuela. trinaco@cantv.net
(3) Laboratorio de Síntesis Orgánica Universidad Central de Venezuela. Caracas, Venezuela. jdomingu@cantv.net
RESUMEN
Se presentan resultados del estudio de modelado molecular de una serie de compuestos del tipo E-1-[4-(3-(sustituido) acriloil)fenil]-3- tosilureas, para las cuales se ha reportado actividad antimalárica como inhibidores del desarrollo in vitro de cepas de P. falciparum resistentes a la Cloroquina. El estudio revela que los compuestos más activos biológicamente muestran una clara superposición en el fragmento comprendido entre el carbono carbonilo del sistema α,β-insaturado y el átomo de azufre del grupo sulfonilo. Igualmente, los resultados señalan que la monosustitución con flúor en posición para y la disustitución con flúor en posiciones 2,4 del anillo B de la chalcona, conducen a un incremento de la actividad; la presencia de tres grupos metoxi y del grupo metilendioxi en el anillo B favorece significativamente la actividad. El estudio teórico incluye cálculos de energías de los orbitales HOMO y LUMO, distancias interatómicas, ángulos rotables, superdeslocalizabilidad, mapas de potencial molecular y otros parámetros electrónicos y moleculares.
Palabras clave: Modelado Molecular; diarilsulfonilureas; malaria
ABSTRACT
Presents the results of the study of molecular modeling a series of compounds of the type E-1-[4-(3-(substituted)acryloyl)phenyl]-3- tosylureas, for which has been reported antimalarial activity as inhibitors of the development in vitro of strains of P. falciparum resistant to chloroquine. The study reveals that the most active compounds biologically show a clear overlap in the fragment between the system carbonyl carbon a,β- unsaturated and the group sulfonyl sulfur atom. Also, the results indicate that monosubstitution with fluoro in para position and disustitucion with fluoro in the B ring of the chalcone 2,4 positions, leading to an increase in activity; the presence of three methoxy groups and the group methylenedioxy in the ring B significantly enhances activity. The theoretical study includes calculations of energies of the HOMO and LUMO orbitals, interatomic distances, angles rotatable, superdeslocalizability, molecular potential maps and other electronic and molecular parameters.
Key words: Molecular Modeling; diarylsulfonylureas; malarie
INTRODUCCIÓN
Las sulfonilureas son compuestos con amplia y variada actividad biológica que incluye entre otras, la antidiabética (Yngve, 1985), oncolítica, la antineoplásica (Mohamadi, Spees y Grindey,1992; Boder y Ehlhardt,1992), diuréticas (Delarge y Lapiere, 1980) y antimalárica (León y otros, 2007). Algunos estudios indican que ciertas drogas antimaláricas podrían afectar el metabolismo del azúcar (Skinner-Adams y Davis, 1999) y otros trabajos sugieren que las sulfonilureas poseen grupos farmacofóricos con propiedades antimaláricas, que podrían presentar un efecto adicional cuando se combinan con el núcleo a,b-insaturado de las cetonas (Domínguez y otros, 1997).
El interés en esta nueva familia de compuestos se ha incrementado, y en la última década trabajos 3D-QSAR han permitido proponer para las sulfonilureas modelos de actividad anticancerígeno entre otras; a partir de esta premisa se ha planteado la síntesis de diarilsulfonilureas como moléculas óptimas (Howert y Grossman, 1990; Gil y otros, 1999). En este estudio se emplean técnicas de modelado molecular para estudiar algunos patrones estructurales y parámetros moleculares, de un grupo de diarilsulfonilureas sintéticas, para las cuales se evaluó su actividad antimalárica, a fin de hallar propiedades que pudiesen estar relacionadas con la actividad biológica reportada (León y otros, 2007).
MÉTODO
Parte Experimental Modelado Molecular
Simulaciones
Todos los cálculos fueron realizados en una estación de trabajo CAChe utilizando licencia de Software de la compañía Fujitsu Limited, CAChe Scientific, versión 4.9. La Mecánica Molecular utilizada por el sistema CAChe emplea el campo de fuerza MM2 de Allinger (Allinger, 1977; Seibel y Kollman, citado por Hansch, Sammes y Taylor, 1990), mejorado por CAChe Scientific, versión 4.9 (CAChe, 2000-2002). A partir del editor del sistema CAChe se construyeron los modelos tridimensionales de los compuestos a-n, con la mejor aproximación de sus distancias internucleares, anillos, ángulos de enlaces, configuraciones atómicas y valencias. La configuración que se asignó al átomo de azufre en el núcleo sulfonilurenilo, fue dxysp3, tomando como referencia la configuración que tiene este átomo en la molécula del Piroxicam que aparece en la base de datos de la estación de trabajo. Posteriormente estas estructuras fueron optimizadas mediante Mecánica Molecular (MM), a partir del método de Block Diagonal Newton Rapson en 600 pasos o hasta alcanzar la convergencia de 0,00100 Kcal/mol.
Para cada una de las estructuras se determinaron luego las conformaciones más estables a partir de cálculos de Dinámica Molecular (DM), calentando virtualmente las moléculas a una temperatura de 900 K, y midiendo la energía asociada a cada conformación cada 1,0000 ps por Block Diagonal Newton Rapson. Seguidamente, se realizó un análisis de las conformaciones de cada compuesto y se seleccionaron los confórmeros energéticamente más estables para cada estructura con una diferencia de 5 Kcal/mol. Las conformaciones de menor energía obtenidas de los cálculos de Dinámica Molecular fueron optimizadas por MM. La conformación más estable fue posteriormente sometida a una búsqueda geométrica alrededor del ángulo t1 (figura 1). La búsqueda geométrica se realizó rotando el ángulo cada 30°, entre 0° y 360°.
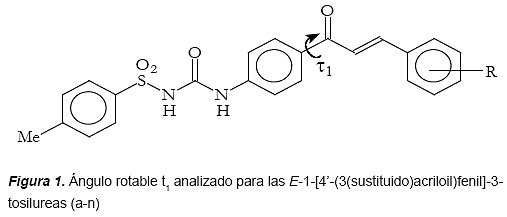
Los diagramas energéticos fueron analizados y como resultado se identificaron las conformaciones más estables que diferían en 5 Kcal/mol de la conformación de mínima energía.
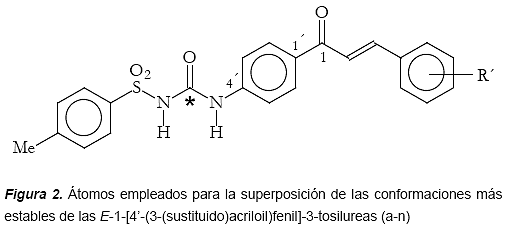
Superposición de las moléculas Una vez obtenidas las conformaciones de mínima energía para los compuestos a-n, se procedió a realizar la superposición entre las moléculas. La superposición de las estructuras, se hizo tomando como referencia los resultados de sus respectivas actividades biológicas. Primero se agruparon aquellos compuestos que resultaron muy activos como inhibidores del desarrollo de cultivos de P. falciparum (IC50 = 1μM) y se superpusieron entre sí (figura 3). Luego se tomó el compuesto activo (b) como referencia para la superposición y los compuestos poco activos (IC50 > 10μM) fueron alineados con respecto a éste (figuras 4 y 5). Tomando como referencia un trabajo relacionado (Michaux y otros, 2001), para la superposición de los compuestos se seleccionaron los átomos C1´, C4´, el carbono carbonilo del núcleo urenilo (C*) y el átomo de azufre del grupo sulfonilo (figura 2).
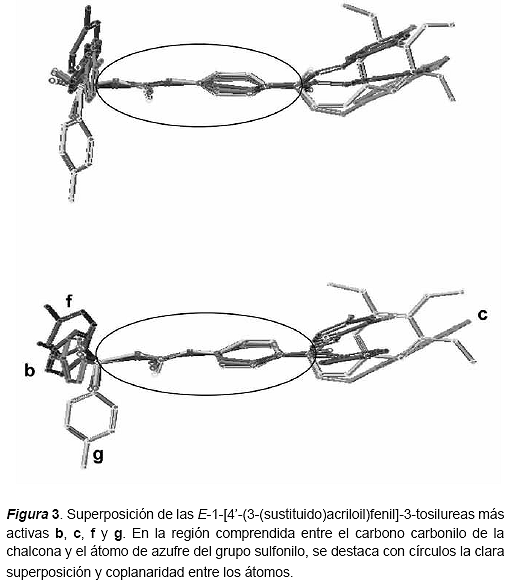
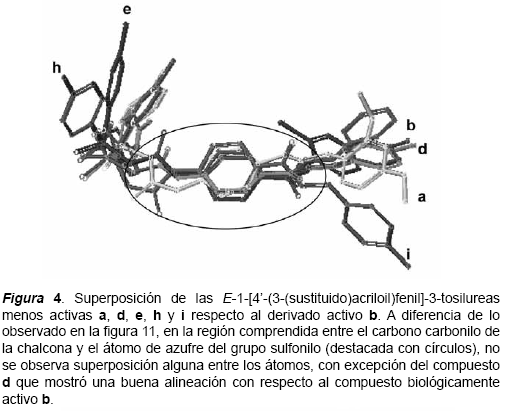
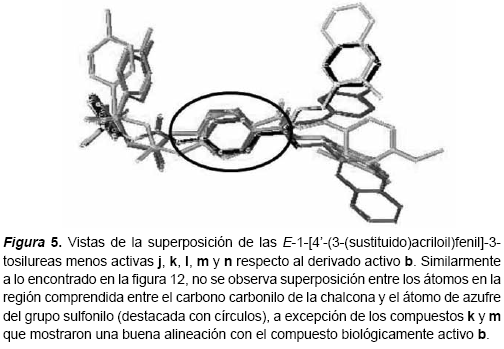
Como resultado de estas superposiciones, se observó que los compuestos activos guardan una clara coplanaridad entre los átomos del fragmento definido por el carbono carbonilo del sistema α,β-insaturado y que se extiende hasta el grupo sulfonilo. Los valores de RMS obtenidos (0,007294; 0,0027195; 0,065600) indican un alto grado de superposición entre los compuestos b-c, bc-f, bcf-g respectivamente. Por el contrario, en los casos de los compuestos poco activos esta coplanaridad no se observó; solamente en los casos de los compuestos d, k y m se observó una importante aproximación a la coplanaridad observada en los compuestos activos.
Cálculos de distancias interatómicas
Con base en los resultados de la superposición para los compuestos a-n, a partir de las conformaciones de mínima energía, fueron calculadas las distancias interatómicas entre los átomos C1-C*, C1-N* y C1-S. Adicionalmente se definió un centroide ![]() en el anillo aromático B del núcleo de la chalcona en cada uno de los derivados y posteriormente también fueron calculadas las distancias entre este centroide y el átomo de azufre en el grupo sulfonilo (Ver cuadro 1).
en el anillo aromático B del núcleo de la chalcona en cada uno de los derivados y posteriormente también fueron calculadas las distancias entre este centroide y el átomo de azufre en el grupo sulfonilo (Ver cuadro 1).
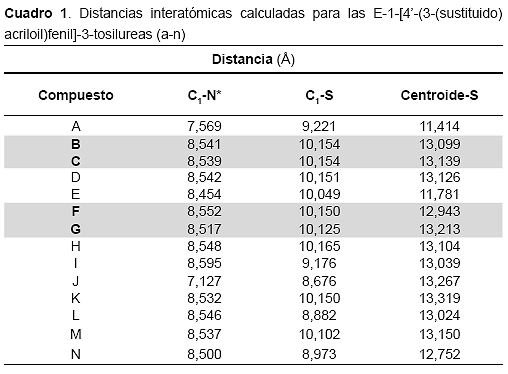
Resulta interesante que en los casos de los derivados activos b-c y f-g, se observa una gran similitud entre las distancias calculadas. En los casos de los compuestos poco activos d, e, h, k y m también se observó que las distancias calculadas guardan estrecha relación con respecto a las de los compuestos activos, mientras que en el resto de los compuestos, se aprecian diferencias significativas entre las distancias calculadas, respecto a las de los compuestos b-c y f-g.
Cálculo de Propiedades Electrónicas de las E-1-[4-(3-(sustituido)acriloil) fenil]-3-tosilureas (a-n).
Las conformaciones de mínima energía obtenidas fueron utilizadas para realizar el resto de los cálculos de Mecánica Cuántica, para lo cual se empleó el programa MOPAC 2002 versión 4.9. Las propiedades electrónicas calculadas fueron visualizadas utilizado el Tabulador del sistema CAChe, con el propósito de convertir los datos en coordenadas en las tres dimensiones y obtener los gráficos correspondientes a los orbitales HOMO y LUMO, así como también sus respectivas energías, densidad electrónica y superdeslocalizabilidad.
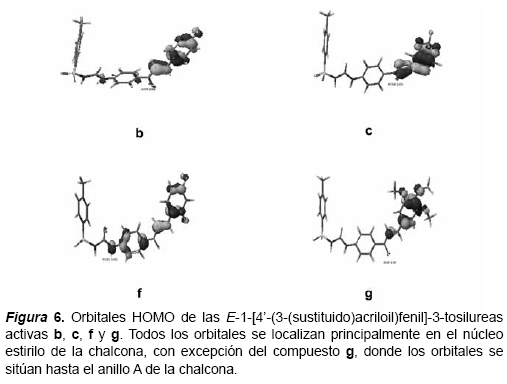
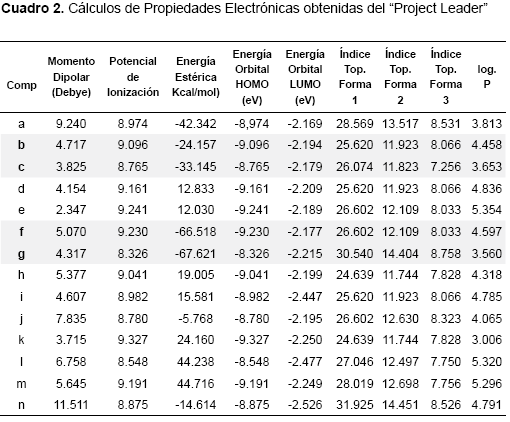
El gradiente de densidad proporciona información acerca de las zonas de alta y baja probabilidad de encontrar una mayor densidad electrónica en la estructura. La figura del orbital HOMO indica la superficie de densidad electrónica con los sitios más reactivos, basado en la distribución de electrones del orbital LUMO, así como la del orbital LUMO se basa en la distribución de electrones del orbital HOMO. La máxima deslocalización (superdeslocalizabilidad) revela los sitios reactivos sobre la base de la distribución de los electrones en los orbitales de frontera. Adicionalmente, con el empleo del programa CAChe Project Leader se calcularon índices geométricos tal como el índice topológico de forma 1 (Kappa 1), que cuantifica el número de ciclos en una estructura química. También se calcularon los índices topológicos de forma 2 y 3 (Kappa 2 y Kappa 3), en los cuales se cuantifica respectivamente el grado de linealidad de una estructura y el grado de ramificación desde el centro de una estructura química. Otras propiedades calculadas y consideradas en el análisis de la posible relación estructura química actividad biológica fueron el momento dipolar y el log. P o coeficiente de partición teórico octanol/H2O.
En los resultados obtenidos de los cálculos del Project Leader se observa una gran semejanza para la mayoría de los parámetros medidos.
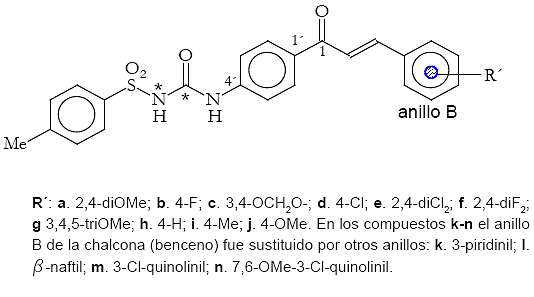
Las semejanzas entre las propiedades electrónicas y la superdeslocalizabilidad determinadas para cada uno de los compuestos biológicamente activos (b-c, f-g) y poco activos (a, d-e, h-n), se verifican en las figuras 6-8.
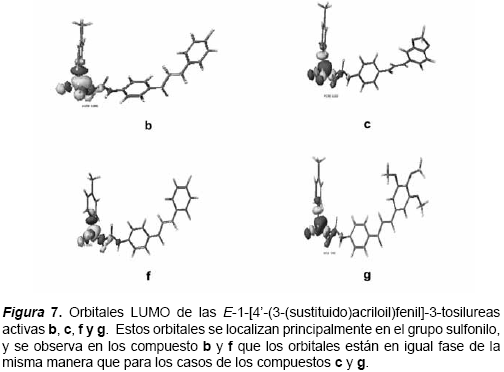
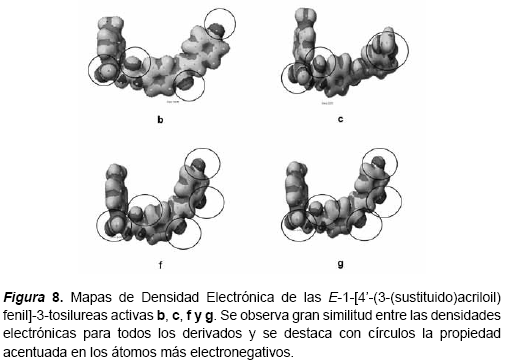
RESULTADOS
A partir de los resultados obtenidos del estudio de modelaje molecular de los derivados de la serie de las E-1-[4-(3-(sustituido)acriloil)fenil]-3- tosilureas (a-n), se aprecia una clara superposición para los compuestos más activos b, c, f, y g, en el fragmento comprendido entre el carbono carbonilo del sistema α,β-insaturado (C1) y el átomo de azufre del grupo sulfonilo, mientras que esta superposición no se observó en los compuestos menos activos de la serie, y esto podría estar relacionado con los resultados obtenidos a partir del ensayo de inhibición del desarrollo de cultivos de P. falciparum.
Por otra parte, aún cuando los compuestos poco activos d, e, h, k y m, guardan similitud con respecto a los compuestos activos b, c, f y g, en cuanto a las distancias calculadas entre los átomos C1-C*, C1-N*, C1-S y Centroide-S, se observan diferencias bastantes grandes entre sus valores de energías estéricas (tabla 2). Esto podría sugerir que para la actividad biológica posiblemente resulte más importante la orientación espacial de los átomos, que las distancias que existe entre estos. Adicionalmente, también se podría explicar por qué a pesar de que los compuestos d, k y m mostraron una buena superposición con el compuesto activo utilizado como plantilla (b), no resultaron biológicamente tan activos como los compuestos b, c, f y g.
Finalmente, la correlación entre los estudios de actividad biológica y la estructura química de los derivados de la serie de las E-1-[4-(3- (sustituido)acriloil)fenil]-3-tosilureas sugiere que la monosustitución en posición para del anillo B con un átomo de flúor conducen a un incremento de la actividad, así como la disustitución en posición 2,4 con este mismo átomo. Además la presencia de tres grupos metoxilos en el anillo B de la chalcona favorece significativamente la actividad, al igual que la presencia del grupo metilendioxi. Esto está de acuerdo con las semejanzas cualitativamente observadas en los mapas de densidad electrónica y de máxima deslocalización que fueron encontrados para los compuestos b, c, f y g.
CONCLUSIONES
El estudio de modelado molecular de los derivados de las E-1-[4-(3- (sustituido)acriloil)fenil]-3-tosilureas, revela que los compuestos b, c, f, y g muestran una clara superposición en el fragmento comprendido entre el carbono carbonilo del sistema α,β-insaturado y el átomo de azufre del grupo sulfonilo; esta disposición espacial de los grupos no se observa en el resto de los compuestos de la serie, y esto posiblemente esté relacionado con los resultados obtenidos a partir del ensayo de inhibición del desarrollo de cultivos de P. falciparum.
La correlación entre los estudios de actividad biológica y la estructura química de los derivados de la serie de las E-1-[4-(3-(sustituido)acriloil) fenil]-3-tosilureas sugiere que la monosustitución en posición para del anillo B con un átomo de flúor conducen a un incremento de la actividad, así como la disustitución en posición 2,4 con este mismo átomo. También la presencia de tres grupos metoxilos en el anillo B favorece significativamente la actividad, así como la presencia de otros grupos sustituyentes que también contienen átomos de oxígeno, tal como el grupo metilendioxi. Esto está en concordancia con las semejanzas cualitativamente observadas en los mapas de densidad electrónica y superdeslocalizabilidad de los compuestos b, c, f y g.
Con base en los resultados obtenidos en esta investigación, se recomienda continuar con la síntesis de otros compuestos relacionados a las N-chalconas, haciendo modificaciones en el patrón de sustitución del anillo A, además de la sustitución en los anillos B y C, a fin de establecer como estos cambios estructurales pueden influir sobre la actividad biológica.
Los resultados de esta investigación sugieren que las E-1-[4-(3- (sustituido)acriloil)fenil]-3-tosilureas podrían perfilarse como buenos inhibidores del desarrollo in vitro de cultivos de P. falciparum, agente causal de la malaria, por cuanto se encontró una buena correlación entre los estudios teóricos y los resultados experimentales, lo cual representa un aporte importante en la búsqueda de nuevas moléculas de candidatos con potencial actividad antimalárica.
Actualmente el modelaje molecular representa una técnica computacional de gran utilidad para estudiar distintos arreglos y parámetros electrónicos/moleculares de diferentes estructuras, y para predecir/ establecer el comportamiento químico/biológico de posibles moléculas de candidatos, lo cual la convierte en un poderoso recurso de apoyo para la docencia y la investigación de estudiantes de química, biología, farmacia y áreas relacionadas.
Este trabajo ha sido financiado por la Universidad Central de Venezuela, y la Universidad Pedagógica Experimental Libertador mediante el Proyecto CICNAT No. 09066, donde participan los autores de este documento.
REFERENCIAS
1. Allinger, N. (1977). Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc., 99, 8127-8134 [ Links ]
2. Boder, G. y Ehlhardt, W. (1992). Discovery of novel antineoplastic sulfonylureas. Drugs Future. 17, 1111-1114. [ Links ]
3. CAChe (Computer Aided Chemistry) (2000-2002). Manual de referencia. Fujitsu Limited. CAChe Scientific, Beaverton Oregon. E.E.U.U. Versión 4.9 [ Links ]
4. Delarge, J. y Lapiere, C. (1980). Synthése et propietés diurétiques de quelques sulfonylurées apparentées au torasémide. Eur. J. Med. Chem. 15, 299-304. [ Links ]
5. Gil, M., Manú, M., Martínez, V., González, A., Encío, I., Arteaga, C., Migliaccio, M. (1999). New anti-neoplastic agents: design, synthesis and experimental determination. ANALES. Sistema Sanitario de Navarra, Departamento de Salud del Gobierno de Navarra. 22, suplemento 3. [ Links ]
6. Howert, J. y Grossman, C. (1990). Novel agents effective against solid tumors: The diarylsulfonylureas. Synthesis, activities, and analysis of quantitative structureactivity relationships. J. Med. Chem. 33, 2393- 2407. [ Links ]
7. León, C., Rodrigues, J., Gamboa de Domıínguez, N., Charris, J., Gut, J., Rosenthal, Ph. y Domínguez, J. (2007). Synthesis and evaluation of sulfonylurea derivatives as novel antimalarials. Eur. J. Med. Chem., 42, 735-742. [ Links ]
8. Michaux, C., Rolin, S., Dogné, J., Durant, F., Masereel, B., Delarge, J. y Wouters, J. (2001). Structure determination and comparison of BM567, a sulfonylurea, with terbogrel, two compounds with dual action, thromboxane receptor antagonism and thromboxane synthase inhibition. Bioorg. Med. Chem. Lett., 11, 1019-1022. [ Links ]
9. Mohamadi, F., Spees, M. y Grindey, G. (1992). Sulfonylureas: A new class of cancer chemotherapeutic agents. J. Med. Chem. 35, 3012- 3016. [ Links ]
10. Seibel, G. y Kollman, P. (1990). Molecular Mechanics and the modeling of drug structures. Comprenhensive Medicinal Chemistry. Vol. 4. Quantitative Drug Design. Citado por Hansch, C., Sammes, P., Taylor, J. and Ramdsen, C., Ediciones Pergamon, 125-138. [ Links ]
11. Skinner-Adams, T. y Davis, T. (1999). Synergistic in vitro antimalarial activity of omeprazole and quinine. Antimicrob. Agents Chemother. 43, 1304-1306. [ Links ]
12. Yngve, M. y Mikael, D. (1985). Process for the preparation of a pharmacologically active sulphonylurea derivative. Patent EP0149592. [ Links ]
