










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalus
versión impresa ISSN 1316-7138
Salus vol.16 no.3 Valencia dic. 2012
Concentración de inhibidores anti VIII en pacientes con hemofilia A que acudieron a una consulta hematológica
Judith Bimanis, María Rojas, Melissa Tovar, Heiddy Vargas
Unidad Bioanalítica de Investigaciones Hematológicas (UBIH). Facultad de Ciencias de la Salud. Universidad de Carabobo Correspondencia: Judith Bimanis Email: judithlab7@gmail.com
RESUMEN
El desarrollo de inhibidores anti factor VIII de la coagulación sanguínea es el mayor problema en las complicaciones inherentes al tratamiento de los pacientes con hemofilia A. Este estudio estuvo dirigido a establecer la frecuencia de casos con inhibidor del factor VIII en un grupo de 41 pacientes con hemofilia A que asistieron a la consulta hematológica de un Banco de Sangre, y los factores asociados al nivel de respuesta del inhibidor anti VIII.
Se utilizó el método de coagulación para la cuantificación del factor VIII, y ensayo Bethesda para la determinación del inhibidor. Los resultados del estudio mostraron que existió mayor prevalencia de hemofilia A severa (53,7%) seguido del grado leve (34,2%) y el moderado (12,2%). De los pacientes estudiados 27 presentaron inhibidor anti VIII y de este grupo 92,6 % eran de baja respuesta (< 5 UB/L), detectándose con mayor frecuencia en casos con hemofilia severa (36,6%), así como también en aquellos pacientes que tenían un rango de edad entre 1 y 10 años (31,7%) y que habían utilizado de manera continua el factor VIII en dosis inferiores a 1000 UDS, sin encontrar diferencias significativas con dosis mayores utilizadas. Por otro lado, la hemartrosis fue la manifestación clínica más frecuente en los hemofílicos (62,8%). Se concluye que el desarrollo de inhibidores de baja respuesta estuvo relacionado fundamentalmente con la gravedad de la enfermedad, con la edad y con el tiempo de exposición al factor VIII de reemplazo.
Palabras clave: Hemofilia A, factor VIII, inhibidor anti VIII, ensayo Bethesda.
ABSTRACT
Anti-factor VIII inhibitor concentration in patients with hemophilia A attending a hematology clinic
The development of blood clotting anti-factor VIII inhibitors is a major problem in the treatment-related complications for hemophilia A patients. This study was designed to establish the frequency of cases with factor VIII inhibitor in a group of 41 patients with hemophilia A attending a Blood Banks hematology clinic, and the factors associated with the response rate of the anti-factor VIII inhibitor. The coagulation method was used for factor VIII quantification, and Bethesda assay for inhibitor determination. The study results showed that there was a higher prevalence of severe hemophilia A (53.7%) followed by mild (34.2%) and moderate (12.2%). From the patients studied, 27 presented anti-factor VIII inhibitor, and from this group 92.6% were low responders (<5 UB / L), more frequently in cases with severe hemophilia (36.6%) and in patients with an age range between 1 and 10 years (31.7%) who had continuously used the factor VIII at doses lower than 1000 UDS. No significant differences were found with higher doses. On the other hand, hemarthrosis was the most common clinical manifestation observed in hemophiliacs (62.8%). It is concluded that the development of low response inhibitors was related primarily to the severity of the disease, age, and time of exposure to factor VIII replacement.
Key words: Hemophilia A, factor VIII, anti VIII inhibitor, Bethesda assay.
Recibido: Febrero 2012 Aprobado: Junio 2012
INTRODUCCIÓN
La hemofilia es un trastorno hereditario caracterizado por la deficiencia funcional o cuantitativa de un factor de la coagulación debido a un defecto en los genes que se encuentran localizados en el brazo largo del cromosoma X, que afecta principalmente a los hombres, mientras que las mujeres portan y transmiten la enfermedad. Las manifestaciones clínicas más comunes son sangrados anatómicos con hemorragias en músculos profundos y articulaciones, hematomas espontáneos, lesiones musculoesqueléticas progresivas y debilitantes (1,2).
Hasta el presente se ha clasificado la hemofilia en tipo A y tipo B; en el tipo A se encuentra deficiente el factor coagulante VIII, y en el tipo B está deficiente el factor IX. Por otro lado, la hemofilia A se presenta en 80 % de los casos de hemofilia, en contraste con 20 a 25 % de casos con Hemofilia B. La severidad de la hemofilia puede clasificarse, de acuerdo al nivel de deficiencia del factor VIII que presente el individuo, en leve con 5-30% del factor, moderada de 1-5% del factor, y grave o severa con un índice inferior a 1% del factor deficiente. Este último grupo es el que con mayor frecuencia presenta manifestaciones clínicas tales como hemorragias espontáneas y hemartrosis (3,4).
El factor deficiente en la hemofilia A es una glicoproteína de síntesis fundamentalmente hepática, que se encuentra en el plasma en muy baja concentración y unido al factor Von Willebrand. La deficiencia cuantitativa o cualitativa del factor VIII: C o IX: C se traduce como defecto en los mecanismos de activación de la coagulación, ya que normalmente el FIX: C y el FVIII: C participan de manera conjunta integrando un poderoso complejo de activación del FX: C, denominado complejo (Xasa). Estos dos factores participan en una fase de sostén o mantenimiento de la coagulación después que el complejo FVII: C/factor tisular ha iniciado la activación del FX:C y FIX:C. La deficiencia de estos dos factores se traduce en un retardo en la formación del coágulo, debido a que la fase inicial de activación de la coagulación dependiente del complejo FVII: C/factor tisular se encuentra suprimida, lo que provoca que la formación del polímero de fibrina se efectúe de manera tardía y clínicamente predisponga al enfermo a hemorragias (5).
Para controlar estos episodios hemorrágicos y compensar la deficiencia de los factores se proporciona un tratamiento cuyo objetivo es incrementar el nivel plasmático del factor deficiente. El tipo de tratamiento que recibe el paciente depende de la gravedad de la enfermedad, de la presencia de inhibidores y del tipo de hemorragia. Los pacientes hemofílicos son tratados con reemplazo del factor de la coagulación, el cual se obtiene del plasma de donantes o puede elaborarse mediante tecnología recombinante (6,7).
El uso del tratamiento sustitutivo puede tener tres vertientes: Una, como tratamiento preventivo o profiláctico primario, el cual se aplica a pacientes con hemofilia severa y se inicia antes o después del primer evento hemorrágico; éste consiste en administrar una o dos veces por semana 20-50 U/Kg/ dosis, y en algunos protocolos cada tercer día. El objetivo es mantener las concentraciones del factor deficiente por encima de 1UI/dl, prevenir oportunamente las hemorragias y limitar las complicaciones tardías de artropatías en esta población. En segundo lugar, como tratamiento profiláctico secundario, donde se colocan dosis preventivas entre 30- 50 U/Kg/dosis antes de un procedimiento odontológico, rehabilitación física o intervención quirúrgica, en cuyo caso la dosis se ajusta al protocolo de acuerdo a las circunstancias. En tercer lugar, como tratamiento sustitutivo, se utiliza de acuerdo a la demanda cuando ocurre un evento hemorrágico.
Por regla general la infusión de 1 UI de Factor VIII por Kg de peso corporal incrementa la concentración de factor VIII en plasma a 2 UI/dl con una vida media de 8-12 horas (8). Este tratamiento puede tener como complicación el desarrollo de inhibidores, es decir, anticuerpos anti VIII y anti IX de clase IgG, principalmente IgG4 de tipo kappa, que se adhieren al factor VIII o IX externos presentes en el tratamiento de reemplazo. La presencia de inhibidores complica el tratamiento de la hemofilia A o B, pues su aparición trae como consecuencia que el nivel de hemorragia en la mayoría de los casos no se logra corregir, o requiere la administración de otros tratamientos tales como factor VII recombinante, y complejo protrombinico activado (5,8).
Otros tratamientos que se utilizan en los pacientes hemofílicos son el crioprecipitado. Éste se prepara mediante el descongelamiento lento de plasma fresco congelado (PFC) a una temperatura de 4° C durante 10 a 24 horas. Contiene cantidades importantes de FVIII (cerca de 5 UI/ ml), factor Von Willebrand (FvW), fibrinógeno y FXIII (pero no FIX o XI). Adicionalmente, también se utiliza el plasma fresco congelado (PFC), que es fuente de todos los factores de coagulación.
La Federación Mundial de la Hemofilia (FMH) apoya el uso de concentrados de factor de coagulación en vez de crioprecipitado o el plasma fresco congelado. No obstante, la FMH reconoce la realidad de que estos tratamientos todavía son ampliamente usados en países de todo el mundo cuando no hay otra opción de tratamiento asequible. (9)
Por otro lado, de acuerdo a las estadísticas reportadas por la Federación Mundial de la Hemofilia, para el año 2009, en Venezuela habían 1.999 personas con hemofilia, de las cuales 1.567 padecían hemofilia A y 432 hemofilia B. Del total de pacientes con hemofilia A, 57 (3.6%) reportaron presencia de inhibidores anti VIII, y en los pacientes con hemofilia B sólo 3 (0.7%) reportaron presencia de inhibidores. Con respecto al tratamiento, Venezuela reportó a la Federación Mundial de la Hemofilia en el año 2.009 que 29 % de los pacientes con hemofilia A recibió factor VIII derivado del plasma y 71% recibió factor VIII recombinante (10).
Aunado a lo expuesto anteriormente, es importante destacar que en la consulta hematológica del Banco de Sangre de la CHET se atienden aproximadamente 120 pacientes hemofílicos de los cuales 83 % corresponde a pacientes con hemofilia A y 17 % a pacientes con hemofilia B.
Con respecto al esquema de tratamiento que reciben, cuando los pacientes ingresan por primera vez al centro médico y no tienen estudios previos que demuestren la carencia de algún factor de la coagulación, es posible que reciban cantidades importantes de crioprecipitado o plasma fresco congelado para corregir el episodio hemorrágico. Sin embargo, una vez que los estudios de laboratorio orientan el diagnóstico hacia una hemofilia tipo A, comienzan a recibir factor VIII derivado del plasma, principalmente el producido por la empresa Venezolana Quimbiotec, o Factor IX de origen plasmático, en el caso de hemofilia tipo B.
Aunque la prevalencia de inhibidores reportados por Venezuela fue baja, existen estudios donde aproximadamente 30% de pacientes con deficiencia del factor VIII y 4% en hemofilia B presentaron inhibidores (11,12). Adicionalmente, otros estudios evalúan el desarrollo del inhibidor del factor en recién nacidos hemofílicos severos expuestos a tratamiento de FVIII recombinante y la relación que existe entre el inhibidor y el efecto de otras variables genéticas y ambientales, donde concluyen que la presencia de un defecto molecular es la variable más importante que influye en el desarrollo del mismo (6); igualmente, otras investigaciones han analizado los factores relacionados al desarrollo de inhibidores del factor VIII en pacientes con hemofilia A, reportando que la primera administración de factor por más de 5 días y la condición severa o moderada del paciente, se asocian principalmente al desarrollo del inhibidor (13).
El objetivo del trabajo fue conocer los factores que se asocian con la formación de inhibidores anti VIII, con el fin de tomar medidas que permitan minimizar la aparición de los mismos, así como también proporcionar datos estadísticos locales que sirvan de referencia a las instituciones de salud regionales, ya que en el Estado Carabobo no se habían realizado estudios de inhibidores en pacientes hemofílicos.
MATERIALES Y MÉTODOS
Se trató de una investigación de tipo no experimental, trasversal y descriptiva; la muestra estuvo conformada por 41 pacientes del sexo masculino con hemofilia A, en edades comprendidas entre 1 y 65 años de edad, que asistieron a la consulta hematológica del Banco de Sangre de la Ciudad Hospitalaria Dr. Enrique Tejera, ubicada en el Edo. Carabobo, entre enero y junio de 2011. Se realizó una revisión de la historia clínica, con el fin de recolectar datos como la edad, el tratamiento recibido en los últimos dos años, la dosis utilizada, y las manifestaciones clínicas más frecuentes. Se cuantificó la concentración del factor VIII de la coagulación y la concentración del inhibidor anti VIII, en caso de presentarlo.
Fueron excluidos del estudio los pacientes que recibieron tratamiento con el factor las 2 semanas previas a la toma de la muestra. Se obtuvo el permiso de la dirección del Banco de Sangre de la CHET y de la Coordinación Regional del Banco de sangre Carabobo Dr. Lorenzo Hands para tener acceso a dicha información; posteriormente se obtuvo el consentimiento informado por escrito de cada individuo o de su representante legal para participar en la investigación.
Cuantificación del factor VIII. Se utilizó el método de referencia que sugiere la Federación Mundial de la Hemofilia para cuantificación de factores, el cual se fundamenta en los tiempos de coagulación de una serie de diluciones de un plasma de referencia (pool) a las que se les añade substrato deficiente del factor en estudio y la tromboplastina; estas diluciones se utilizan para realizar una curva estándar en las que se interpolaron los resultados de la muestra problema tratada en las mismas condiciones.
Metodología. 1-Usando volúmenes de 0,2 mL, se hicieron diluciones 1:2 de plasma estándar y plasma de prueba en buffer Tris desde 1/10 hasta 1/40 en tubos de plástico. 2- Se pipeteó 0,1 mL de cada dilución estándar en un tubo de vidrio de 75 × 10 mm. 3- Se Añadió 0,1 mL de plasma deficiente en factor VIII y transfirió a un baño de María a 37 ºC.4- Se Añadió 0,1 ml de reactivo de TTPA e incubó durante 3 minutos. 5- A los 3 minutos, se añadió 0,1 mL de CaCl2 y se registró el tiempo de coagulación. 6- Finalmente se representaron los resultados sobre una hoja para escala logarítmica/lineal. A la dilución 1/10 se le asigna arbitrariamente un valor de 100%; a la dilución de 1/20, un valor de 50% y a la dilución de 1/40, un valor de 25%.
Se realizó la lectura de la concentración de la muestra de prueba interpolando los resultados en la curva estándar. De esta forma se obtuvo la concentración en porcentaje de actividad o en unidades de actividad. (14-16).
Ensayo Bethesda. El ensayo Bethesda permitió demostrar la presencia de inhibidores anti VIII. El mismo se fundamenta en que al mezclar partes iguales del plasma normal y plasma del paciente e incubarlo por 2 horas a 37ºC se demuestra la presencia de inhibidores cuando el factor VIII residual disminuya a menos de 50%. (17-20) El método utilizado se describe a continuación: 1- Se prepararon diluciones usando tampón de ensayo de factor VIII del plasma del paciente. 2- Se agregó una cantidad estandarizada de factor VIII en forma de pool de plasma normal a cada dilución de plasma de prueba. Así, cada mezcla para incubación tiene una concentración inicial de aproximadamente 50 U/dL. No es importante una concentración precisa, porque se agrega el mismo pool de plasma normal a todas las mezclas para incubación. 3- En el ensayo de factor VIII que se practica después de transcurridas las dos horas de incubación, la mezcla de control de plasma normal y plasma deficiente en factor VIII se utiliza como referencia estándar, y la concentración de factor VIII de las demás mezclas se calcula comparándola con aquellas, midiendo el nivel residual de factor VIII y calculando el inhibidor a partir de un gráfico de factor VIII residual vs. las unidades de inhibidor. Al final del período de incubación, se mide el nivel residual de factor VIII y se calcula el inhibidor a partir de un gráfico de factor VIII residual vs. las unidades de inhibidor (14).
Análisis Estadístico. Para el análisis estadístico de los resultados se utilizó el paquete estadístico Statistics 18. Los resultados se presentan en tablas de frecuencias para establecer asociación entre nivel de concentración del inhibidor y el grado de severidad de la hemofilia (leve, moderada, severa), la edad, manifestaciones clínicas frecuentes, tratamiento y dosis recibida. Se utilizó la prueba del Chi cuadrado (X2), asumiendo para todas las pruebas un nivel de significancia de p <0,05.
RESULTADOS
La muestra estudiada estuvo conformada por 41 personas del sexo masculino en edades comprendidas entre 1 y 65 años (media: 19,7 años con una desviación estándar = 16,6), con una frecuencia de 22 casos (53,6 %) con hemofilia de grado severo, 14 casos (34,2 %) de hemofilia grado leve y solo 5 casos (12,2%) con hemofilia de grado moderado. En la tabla 1 se pueden observar las diferentes manifestaciones clínicas que presentaron los pacientes (hemartrosis, hematomas, hematuria, hemorragias de diversas causas, dolor articular).
En 7 pacientes no se reportó ninguna manifestación clínica en los últimos dos años. Por otro lado, se aprecia que todos los pacientes recibían Factor VIII en dosis que variaron desde 500 hasta 5000 UDS, algunos con antecedentes de otros tratamientos como plasma fresco congelado (PFC) y crioprecipitado (Criop).

En la Tabla 2 se muestra la presencia del inhibidor anti VIII según el grado de hemofilia, en donde se puede observar que del total de 41 pacientes, solo en 27 casos se encontró presencia de inhibidor anti VIII por método Bethesda, encontrándose la mayor prevalencia en los hemofílicos severos (n: 15; 36,6%), pero sin diferencias estadísticamente significativas (χ2= 1,276; p< 0,05).

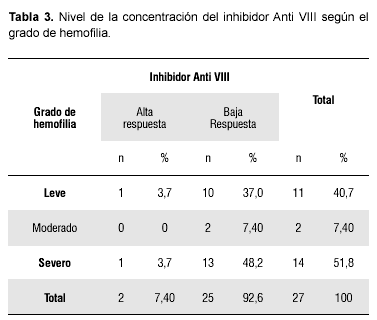
La Tabla 3 presenta la concentración del inhibidor anti VIII de acuerdo al grado de severidad de la hemofilia, donde se observan 2 casos con inhibidor de alta respuesta (mayor de 5 UB) y 25 casos con inhibidor de baja respuesta (menos de 5 UB), con mayor prevalencia en los pacientes con hemofilia severa. Sin embargo, no se evidenció asociación significativa entre la concentración del inhibidor y el grado de hemofilia (χ2= 0,22; p< 0,05).
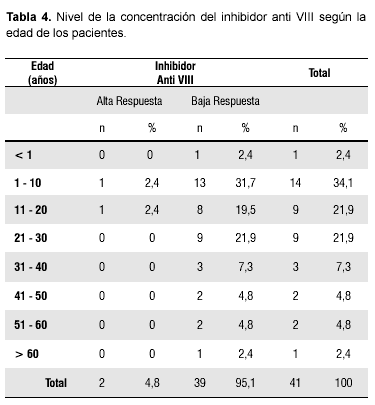
La Tabla 4 representa el nivel de concentración de inhibidor anti VIII según la edad, donde destaca que la prevalencia de inhibidor anti VIII de baja respuesta fue mayor en el grupo con intervalo de edad comprendido entre 1 y 10 años (n=13; 31,7%). No se encontró asociación significativa entre la concentración del inhibidor y los diferentes grupos etarios. (χ2= 0,76; p> 0,05).
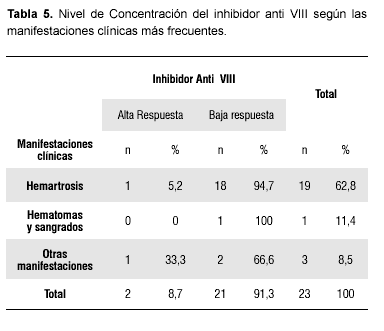
La tabla 5 presenta la relación entre nivel de concentración del inhibidor anti VIII y las manifestaciones clínicas más frecuentes. En base al total de pacientes con presencia de inhibidor, solo 4 casos (0,54 %) no reflejaron manifestaciones recientes en la historia clínica. La manifestación clínica más frecuente (n=19; 62,8%) entre los pacientes con presencia de inhibidor anti VIII fue la hemartrosis, seguido de los hematomas y otras manifestaciones tales como hematuria, dolores articulares y sangrados molares. Todas las manifestaciones clínicas fueron observadas con mayor frecuencia en los pacientes hemofílicos severos y leves, tomando en cuenta que dichas poblaciones (casos severos y leves) son las que tienen mayor representación de individuos. No existió asociación significativa entre la concentración del inhibidor y las diferentes manifestaciones clínicas que presentaron los pacientes. (χ2= 2,67; p> 0,05), aunque en la mayoría de los pacientes con hemartrosis se evidenció anti VIII de baja respuesta.
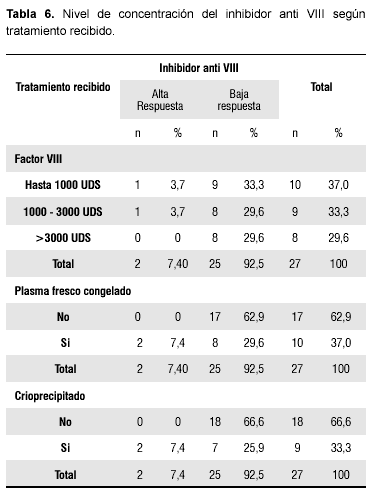
Según las dosis de factor VIII recibidas por los pacientes, predominaron aquéllos que recibieron dosis menores de 1000 UDS; los dos pacientes con niveles altos del inhibidor no superaron los 3000 UDS del factor VIII (Tabla 6). Por otro lado, de los pacientes que desarrollaron inhibidor de baja respuesta, 8 pacientes habían recibido plasma fresco congelado y 7 habían recibido crioprecipitado. Cabe destacar que los dos pacientes que desarrollaron inhibidor de alta respuesta no habían recibido plasma fresco congelado ni tampoco crioprecipitado. No existió asociación significativa entre la concentración del inhibidor y las diferentes dosis de Factor VIII (χ2= 0,91; p> 0,05).
Tampoco existió asociación significativa entre la concentración del inhibidor y los diferentes tratamientos que recibieron los pacientes (χ2= 0,46; p> 0,05).
DISCUSIÓN
La hemofilia tipo A es una de las coagulopatías más frecuentes, en vista de esto resulta importante estudiar los factores que complican el tratamiento de dicha enfermedad. Uno de ellos es la presencia de inhibidores anti VIII. Debido a los escasos reportes sobre tal aspecto en pacientes hemofílicos venezolanos, este trabajo tuvo como objetivo establecer no sólo la frecuencia de casos con inhibidor anti VIII en pacientes hemofílicos que acuden a la consulta hematológica del banco de sangre de la CHET, sino también la asociación entre el nivel de respuesta del mismo y diferentes variables de importancia en la historia médica del paciente.
El porcentaje de pacientes donde se obtuvo un título positivo de inhibidor anti VIII fue de 65,9% (n=27 casos), observando que el mismo es superior al descrito por otros autores, entre ellos Kruce-Jarres (21), quienes indican una frecuencia de casos de aproximadamente 30% de la población en personas con hemofilia A. Sin embargo es importante tomar en cuenta que la muestra estuvo conformada sólo por 41 pacientes hemofílicos. Sería interesante verificar la incidencia y prevalencia de inhibidores en la totalidad de la población de pacientes hemofílicos que son atendidos en la consulta hematológica del Banco de Sangre de la CHET.
Tal como se menciona en relación al título del inhibidor, lo más frecuente es encontrar niveles bajos de anti VIII. En este estudio la gran mayoría (92,6 %) de los pacientes estudiados mostraron niveles de inhibidor menores de 5 UB/L, con un promedio de 0,71 UB/L, es decir, que aunque el estudio demostró la presencia de inhibidores de baja respuesta en un alto porcentaje de la muestra analizada, el nivel del título es muy bajo, no repercutiendo en su clínica y tratamiento.
Queda realizar el seguimiento de estos pacientes con una nueva cohorte para determinar si se trata de anticuerpos transitorios o permanentes. Solo en dos de los pacientes estudiados se demostró la presencia de inhibidor de alta respuesta, se deduce que el desarrollo de niveles altos por lo general se debe a un mecanismo de memoria que se desencadena después de que el paciente ha recibido tratamiento. Por otro lado, Wang et al (7), Kempton et al (22), y Yan et al (23), han señalado que los pacientes que tienen mayor riesgo de desarrollar inhibidores son aquellos con hemofilia severa. En la presente investigación, aunque prevaleció la presencia de inhibidor de baja respuesta entre los pacientes con hemofilia de grado grave, no se demostró asociación estadísticamente significativa entre la presencia del inhibidor y el grado de severidad de la enfermedad. Por otro lado, es interesante destacar que las estadísticas de la consulta hematológica del Banco de Sangre de la CHET, con respecto al total de pacientes hemofílicos que atienden, concuerdan con los resultados del presente estudio en cuanto a la mayor prevalencia de hemofilia de grado severo y leve. Cabe destacar que los pacientes con hemofilia de grado leve reciben el Factor VIII plasmático por traumatismos que sufren, debido a que al pertenecer al grupo de hemofílicos de menor riesgo, piensan que pueden llevar una vida normal y son los que menos cuidados profilácticos aplican en su cotidianidad. Ésta es probablemente una de las causas que influyen en la presencia de inhibidores de baja respuesta en los pacientes con hemofilia leve.
Con respecto a la relación entre el nivel de inhibidor y la edad, la mayor prevalencia de anticuerpos de baja respuesta se observó en el intervalo de edad de niños de 1 a 10 años. En este sentido, estos resultados coinciden con los informados por DiMichelle. (24) y Chalmers et al (25) quienes reseñan que el desarrollo del inhibidor ocurre en una edad promedio entre 1 y 2 años después de un periodo de 10 tratamientos; de forma similar, Kruse-Jarres (21) concluyó que la exposición al factor VIII de reemplazo durante el primer año de vida es una variable que influye notablemente en el posterior desarrollo de inhibidores. También Kempton et al (22) indicaron en su estudio una prevalencia mayor de inhibidor entre los pacientes con edad comprendida entre 2 a 14 años. Estudios como el de DiMichele. (24) y Goudemand et al (26) han demostrado que el riesgo de desarrollar inhibidores aumenta con la mayor exposición a las diferentes formas de tratamiento como crioprecipitado, plasma fresco congelado y Factor VIII recombinante, es decir, que periodos largos de los mismos están asociados con un aumento en el riesgo de desarrollar inhibidores.
En los pacientes con presencia de inhibidor anti VIII, la hemartrosis fue la manifestación clínica más frecuente, evidenciándose en dos de los pacientes con niveles altos del inhibidor; uno que presentó hemartrosis, y el otro, traumatismo en la cabeza. Estos datos coinciden con lo informado por Girolami et al (27), quienes en un grupo de pacientes hemofílicos encontraron el predomino de hemartrosis como manifestación clínica más frecuente, seguida de la presencia de hematomas y otras hemorragias en menores porcentajes. A pesar de que la muestra seleccionada para este estudio fue con pacientes que presentaron inhibidores, no hubo diferencias estadísticamente significativas con respecto al total de pacientes de la consulta en cuanto a las manifestaciones clínicas más frecuentes.
Diversas investigaciones (25, 28) han demostrado una relación entre la presencia de inhibidor anti VIII y las dosis de tratamiento con factor VIII, y también con el hecho de recibir terapia múltiple, es decir, en conjunto con plasma fresco congelado y crioprecipitado y/o factor VIII plasmático o recombinantes alternos. Contrario a lo anterior, en la presente investigación los dos únicos pacientes que desarrollaron inhibidor de alta respuesta no recibieron plasma fresco congelado, ni crioprecipitado ni factor VIII recombinante en los dos últimos años.
Al recibir consecuentemente un solo tipo de tratamiento sustituto del factor VIII se previene la formación de inhibidores que afectan la respuesta al tratamiento y, en caso de desarrollarlos, éstos son de baja respuesta, tal como se evidenció en los resultados del estudio.
Tampoco se demostró una asociación significativa entre la concentración del inhibidor y las diferentes dosis de factor VIII o terapia combinada con plasma fresco congelado o crioprecipitado. Sin embargo, sí puede estar asociado al tiempo de exposición a la terapia de reemplazo, debido a que la mayoría de los pacientes estaban sometidos a largos períodos de tratamiento profiláctico con bajas dosis de factor VIII, a fin de prevenir episodios hemorrágicos en los pacientes hemofílicos severos; y en el caso de los hemofílicos leves, éstos recibieron el factor VIII de acuerdo a la demanda del evento hemorrágico que manifestaron.
Una de las limitaciones importantes de este estudio fue el bajo número de pacientes participantes, debido a que tenían que permanecer dos semanas sin colocarse el factor VIII para la toma de muestra. No obstante, ésta es la primera investigación a nivel regional que muestra datos sobre la presencia del inhibidor anti VIII en pacientes hemofílicos, por lo que se plantea la necesidad de dar continuidad a la investigación con estudios de diseño prospectivo, a fin de completar los datos que permitan conocer con mayor exactitud los factores que influyen en la aparición de anticuerpos en los pacientes hemofílicos venezolanos.
En conclusión, fue frecuente la presencia de inhibidor anti VIII, con títulos menores de 5 UB/L en el grupo estudiado, y los niveles de respuesta del inhibidor no se encontraron asociados a la severidad de la hemofilia, la edad, manifestaciones clínicas, dosis recibida de FVIII o el tipo de tratamiento recibido. Otros estudios deberán confirmar y ampliar los datos presentados. Dada la alta frecuencia de presencia de inhibidor anti VIII evidenciada, se recomienda el seguimiento por parte del facultativo de los niveles de inhibidor en los pacientes hemofílicos cada tres meses posterior al tratamiento, así como educar al paciente sobre los cuidados que él mismo debe tener a fin de minimizar complicaciones que conlleven al uso prolongado de terapias de reemplazo del factor deficiente.
Agradecimientos: al personal que labora en la Unidad Bioanalítica de Investigaciones Hematológicas, al personal que labora en la asignatura Hematología de la Escuela de Bioanálisis, a los profesores Nelina Ruiz y Aldo Reigoza, y finalmente al personal del Banco de sangre de la Ciudad Hospitalaria Dr. Enrique Tejera, especialmente a la Hematóloga, Katiuska Núñez.
REFERENCIAS
1. Turgeon M. Hematología clínica teoría y procedimientos. Editorial El Manual Moderno. México 2006; p.400-406.
2. Roberts H, Hoffman M. Hemofilias A y B. En: Beuther E, Lichtman M, Calles B, Kipps T, Soligsohn J. Williams. Hematología. Editorial Marban Libros. Madrid 2005; p.1639- 1652.
3. Serrano J. Fundamentos y técnicas de análisis hematológicos y citológicos. Editorial Masson. Barcelona 2005; p. 224. [ Links ]
4. Zapata R. Las hemofilias. En: Cueller F, Falabella F. Hematología. 6ta Edición. Editorial Coorporación para Investigaciones Biológicas. Medellín 2004; p. 258-259.
5. Ruiz J. Fundamentos de hematología. 3era. edición. Editorial Panamericana. Buenos Aires. 2003; p.399-419.
6. Fonrodana F, López M, Coagulopatías Congénitas. En: García J, San Miguel J, Sierra J, Urbano A, Vicente V, Vives J, Hematología. Editorial Arán. Madrid. 2003; p. 400-401.
7. Wang X, Zhao Y, Yang R, Wu J, Sun J, Zhang X, Ding Q, Ge H, Wang H. The prevalence of factor VIII inhibitors and genetic aspects of inhibitor development in Chinese patients with hemophilia A. Haemophilia. 2010; 16: 632-639. [ Links ]
8. Ochoa M, Detección de inhibidores en pacientes con hemofilia hereditaria. 2009; 10-16. Disponible en http://hdl. handle.net/123456789/4353 [Consultado el 10 Enero de 2012]
9. Directrices para el tratamiento de la hemofilia. Federación Mundial de la Hemofilia. 2005; 39-43. Disponible en: http://www.wfh.org/3/docs/Publications/Diagnosis_and_Treatment/Guidelines_Mng_Hemophilia_SP.pdf [ Links ]
10. Reporte de la Federación Mundial de la hemofilia año 2009; 16-32. Disponible en: http://www.wfh.org/2/docs/ublications/Statistics/2009_Global_Survey_Report.pdf
11. Pérez R, Ozelo MC, Villaça P, Solano M, Jiménez G, Martínez C, García J, Mendoza S, Rodríguez I, Ruiz A. Diagnosis and treatment of congenital hemophilia with inhibitors a Latin American perspective. Medicina. Buenos Aires. 2008; 68(3): 227-242. [ Links ]
12. Que son los Inhibidores. Federación Mundial de la Hemofi lia 2010; 3-13.Disponible en http://www. wfh.orglindex_ SP.asp?lang=SP.[Consultado el 11 de Agosto del 2011]
13. Kershaw G, Jayakodi D, Dunkley S. Laboratory identifi cation of factor inhibitors: The perspective of a large tertiary Hemophilia Center. Semin Thromb Haemost. 2009; 35 (8): 760-767. [ Links ]
14. Diagnóstico de la Hemofi lia y otros trastornos de la Coagulación. Manual de Laboratorio. Federación Mundial de la Hemofi lia. 2010; 67 y 81. Disponible en: http://www.wfh.org/3/docs/Publications/Diagnosis_and_Treatment/Lab_Manual/Lab_manual2010_SP.pdf
15. Kitchen S, McCraw A. Diagnósticos de la Hemofi lia y otros trastornos de la coagulación. Manual de laboratorio. Comité de Ciencias. Federación Mundial de la Hemofi lia. 2010. P 53-55. 16. Brotons M, Métodos diagnósticos de los trastornos hemorrágicos y control del tratamiento anticoagulante. En: Vives J, Aguilar J. Manual de Técnicas de Laboratorio en Hematología. 2da Edición. Editorial Masson. Barcelona 2001; p. 540-548.
17. Gómez D, Jaime J. Hemostasia y Trombosis en Ruiz G. Fundamentos de interpretación clínica de los exámenes de laboratorio. Editorial Panamericana. Buenos Aires 2004. p. 100-101.
18. Kim Y, Kang Y, Lee W. Comparative measurement of FVIII inhibitors in hemophilia a patients using ELISA and Bethesda assay. Korean J Lab Med. 2010; 30: 260-263. [ Links ]
19. Sahud M, Pratt K, Zhukov K, Thompson R. ELISA system for detection of immune responses to FVIII322. Disponible en http://www.ncbi.nlm.nih.gov/pubmed/17498082 [Consultado el 13 de Noviembre del 2011]
20. Bert V, Waander L, Van H, Britta A, Laros-van V. Improvements in Factor VIII Inhibitors Detection: From Bethesda to Nijmegen. Semin Thromb and Haemost 2009; 35: 752-758. [ Links ]
21. Kruse-Jarres R. Current controversies in the formation and treatment of alloantibodies to factor VIII in congenital hemophilia A. ASH Education Book. 2011; 1: 407-412. [ Links ]
22. Kempton C, Soucie J, Abshire C. Incidence of inhibitors in a cohort of 838 males with hemophilia A previously treated with factor VIII concentrates. J Tromb Haemost. 2006; 4: 2576-2581. [ Links ]
23. Yan Z, Fan L, Li X, Wang X, Hua B, Wang S, Zhao Y. Frequency of factor VIII inhibitors in the patients with hemophilia A and environmental risk factors for inhibitor development. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2009; 31: 580-583. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/19968075 [consultado el 15 de Agosto de 2011] [ Links ]
24. DiMichele, D. Inhibidores en Hemofi lia: información básica. Federación Mundial de la Hemofi lia. 4ta edición. 2004; 7: 1-4. Disponible en: http://www.wfh.org/3/docs/Publications/Inhibitors/TOH-7_Inhibitor-Primer_Rev2008_SP.pdf. [consultado el 15 de Agosto de 2011] [ Links ]
25. Chalmers E, Brown S, Keeling D, Liesner R, Richrds M, Stirling D, Thomas A, Vidler V, Williams M, Young D. Early Factor VIII exposure and subsequent inhibitor development in children with severa hemophilia A. Haemophilia 2007; 13(2): 149-155. [ Links ]
26. Goudemand J, Rothschild C, Demiguel V, Vinciguerrat C, Lambert T, Chambost H. Infl uence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood 2006; 107: 46-51. [ Links ]
27. Girolami A, Vettere S, Ruzzon E, Berti de M, Fabris S. Rare and inosual blending manifestation in congenital bleeding disorders. An annotated review. Clin Appl. Thromb Hemost. 2012; 18:121-127. [ Links ] Epub 2011. Aug. 25.
28. White II, Kentom, Grisley A, Nielsen B, Roberts H. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005; 3(8): 1676-1681. [ Links ]
