










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSalus
versión impresa ISSN 1316-7138
Salus vol.17 no.2 Valencia ago. 2013
Detección de hemoglobinopatías en recién nacidos del Hospital Materno Infantil "Dr. José María Vargas" de la ciudad de Valencia, Venezuela.
Indira Varela1,2, Alida Sequera1, Rhaiza Olivero1
1 Unidad Bioanalítica de Investigaciones Hematológicas (UBIH), Escuela de Bioanálisis, Facultad de Ciencias de la Salud, Universidad de Carabobo. Valencia. Venezuela
2 Departamento de Morfofisiopatología, Escuela de Bioanálisis, Facultad de Ciencias de la Salud, Universidad de Carabobo. Valencia. Venezuela
Correspondencia: Indira Varela. E-mail: indiravarela@cantv.net
RESUMEN
Las hemoglobinopatías constituyen un amplio grupo de desórdenes hereditarios autosómicos recesivos ampliamente distribuidos alrededor del mundo. La población venezolana es una mezcla de tres grandes ramas: indios, españoles y africanos; la presencia de variantes hemoglobínicas, principalmente HbS y HbC, está íntimamente relacionada con la llegada de africanos durante el proceso de la colonización. En el presente estudio se investigó la presencia de variantes de hemoglobina en recién nacidos del Hospital Materno Infantil "Dr. José María Vargas" de la ciudad de Valencia. Se analizaron 507 muestras de sangre de cordón umbilical. A todas las muestras se les practicó electroforesis en acetato de celulosa a pH 8.6; a las muestras que presentaron hemoglobina anormal se les realizó electroforesis en gel de agar a pH 6.5. Para confirmar la presencia de hemoglobinas anormales se realizó estudio de las cadenas de globina empleando HPLC de fase reversa. Del total de muestras analizadas 496 (97.83%) presentaron un fenotipo normal FA; 10 (1.97%) tuvieron un fenotipo FAS; y 1 (0.2%) presentó un fenotipo FAC. Las frecuencias encontradas en este estudio confirman la necesidad de implementar un tamizaje para hemoglobinopatías en la población neonatal y programas de asesoramiento genético.
Palabras clave: hemoglobinopatías, tamizaje neonatal, anemia falciforme, electroforesis, HPLC.
Detection of hemoglobinopathies in newborns
ABSTRACT
Hemoglobinopathies are hereditary autosomic recessive disorders with a high prevalence worldwide. Venezuela is a country with a significant ethnical admixture, (Indian, European, African) in which the colonization process played a great role on the spread of abnormal genes of hemoglobin variants. Specifically, HbS and HbC were introduced mainly by the descendent of African slaves. In the present study we investigated the presence of hemoglobin variants in newborns from the maternity hospital "Dr. José María Vargas", Valencia. Samples of umbilical cord blood from a total of 507 newborns were analyzed. All samples were submitted to cellulose acetate electrophoresis at pH 8.6. Electrophoresis in agar gel at pH 6.5 was performed on samples presenting abnormal hemoglobin. Globin chains were studied using reversed phase HPLC to confirm abnormal hemoglobin. From a total of 507 samples, 496 (97.83%) presented with normal hemoglobin FA; 10 (1.97%) with phenotype FAS and 1 (0.2%) with phenotype FAC. Frecuencies found in the present study confirm the need to implement screening for hemoglobinopathies in the neonatal population, as well as a genetic counseling program.
Key words: Hemoglobinopathies, neonatal screening, sickle-cell anemia, electrophoresis, HPLC.
Recibido: Octubre 2012 Aprobado: Mayo 2013
INTRODUCCIÓN
Las alteraciones de la hemoglobina (Hb) o hemoglobinopatías constituyen un amplio grupo de desórdenes hereditarios autosómicos recesivos que incluyen alteraciones cualitativas o cuantitativas de la globina, secundaria a alteraciones genéticas que condicionan cambios en la estructura o en la síntesis de la molécula de hemoglobina. La sustitución de aminoácidos en el interior de la molécula da lugar a las hemoglobinopatías estructurales mientras que la disminución o ausencia total de síntesis de una o varias cadenas de globina que son estructuralmente normales corres-ponden a las talasemias (1). Aproximadamente 7% de la población mundial es portadora de una mutación potencialmente patológica en uno de los genes de la globina constituyendo uno de los principales problemas de salud pública alrededor del mundo; se calcula que cada año nacen más de 300.000 niños con hemoglobinopatías graves, la mayoría de ellos en países de ingresos bajos y medios (2,3). Las hemoglobinopatías son las enfermedades monogénicas más comunes en algunas poblaciones de África, el área mediterránea, el Caribe, América Central y América del Sur. Las hemoglobinopatías estructurales más amplia-mente extendidas alrededor del mundo son la HbS (África), la HbC (África Occidental), la HbE (Sureste Asiático), y la HbD (Punjab, India) (2). En Venezuela, las variantes estructurales más frecuentes son la S y la C (4-6). La HbS resulta de una mutación puntual en la posición 2 del sexto codón del exón 1 del gen de la betaglobina (beta 6; GAG®GTG) localizado en el cromosoma 11, lo que se traduce en una sustitución de ácido glutámico por valina en la posición 6 de la cadena betaglobina. La HbS puede expresarse bajo 4 formas principalmente: a) Forma heterocigota conocida como rasgo falciforme o rasgo drepanocítico (HbAS); los individuos heterocigotos por lo general son asintomáticos. b) Forma homocigota o anemia de células falciformes (HbSS). c) Doble heterocigoto HbS-talasemia. d) Doble heterocigoto con otras variantes estructurales (HbSC, HbSD, HbSX). Los individuos homocigotos o doble heterocigotos presentarán anemia hemolítica crónica, crisis dolorosas por oclusión de los vasos y elevado riesgo infeccioso por asplenia funcional (7). La HbC, como la HbS, es una variante estructural de la cadena beta de la hemoglobina que resulta de una mutación única en la posición 1 del codón seis del gen beta (beta 6; GAG®AAG) lo cual resulta en la sustitución del ácido glutámico por lisina en la posición seis de la cadena betaglobina. Los individuos homocigotos (HbCC) presentan una anemia hemolítica de leve a moderada; el cuadro clínico se debe a que la HbC induce deshidratación del eritrocito y formación intracelular de cristales. Los dobles heterocigotos SC sufren de anemia grave pero más leve que la anemia falciforme (8). De acuerdo a la OMS (9) la estrategia más rentable para reducir la carga de hemoglobinopatías consiste en combinar el tratamiento con programas de prevención. El propósito de la detección temprana de hemoglobinopatías es la de identificar desórdenes clínicamente importantes y proveer asesoramiento genético, educación y cuidados especiales antes de que se establezcan los síntomas clínicos con el fin de mejorar la calidad de vida y disminuir la mortalidad. Se ha observado que el diagnóstico neonatal de la anemia falciforme puede reducir sustancialmente la morbimortalidad durante los primeros 5 años de vida de los infantes (2). El objetivo de la presente investigación fue determinar la frecuencia de hemoglobinopatías en muestras de sangre de cordón umbilical de recién nacidos, destacando la importancia de su detección en la población neonatal.
MATERIALES Y METODOS
Este estudio fue realizado en 507 muestras de sangre de cordón umbilical de recién nacidos del Hospital Materno Infantil "Dr. José María Vargas" de la ciudad de Valencia, Venezuela, recolectadas durante el periodo 2.008-2.010. El estudio se condujo de conformidad con los lineamientos establecidos por las Normas de Investigación Clínica (10) y fue aprobado por el comité de ética del referido centro y en todo los casos se obtuvo el consentimiento informado de los padres.
La colección de sangre fue realizada por el personal especializado bajo la supervisión de los obstetras encargados del parto de acuerdo con el protocolo de rutina para la tipificación sanguínea de ABO y Rh. Se recolectaron 5 ml de sangre en tubos vacutainerTM con EDTA. Estas muestras fueron lavadas tres veces con solución salina fisiológica al 0.154M a 20ºC, con el paquete resultante se prepararon hemolizados con agua altamente purificada en proporción 1/10, los cuales fueron congelados a -70ºC hasta su procesamiento. Todas las muestras fueron sometidas a electroforesis de hemoglobina en acetato de celulosa utilizando un buffer Tris-EDTA-Borato a pH 8.6. Las muestras que presentaron una variante a pH alcalino se les sometió a electroforesis en agar a pH 6.2 (11).
Los casos que fueron positivos para alguna variante se sometieron, para su confirmación, a cromatografía líquida de alta eficiencia (HPLC) de fase reversa para la separación y estudio de las cadenas de globina (beta-A, delta, alfa, G gamma, A gamma y A gamma T); para ello se aplicó el método de Shelton y col. (12) con modificaciones. Se empleó un equipo HPLC Hewlett Packard modelo 1050 con columna de tipo analítico para fase reversa (Vydac C4) de 250mm x 10mm y 300 Å de poro. Los desarrolladores estuvieron constituidos por mezclas de acetonitrilo, agua y ácido trifluoroacético. La fase móvil consistió de un buffer A con mezcla de acetonitrilo-agua en proporción 60:40 con 0,1 ml de ácido trifluoroacético; el desarrollador B constituido por acetonitrilo-agua en proporción 20:80 con 0,1 ml de ácido trifluoroacético. La velocidad de flujo se estableció en 1 ml/min, el monitoreo se realizó a 220 nm y el tiempo de cromatografía en 80 min. Se utilizaron controles de HbFAS y HbFAC.
RESULTADOS
Del total de las 507 muestras analizadas de sangre de cordón umbilical de recién nacidos 496 (97,83%) presentaron un fenotipo neonatal normal (HbFA). El total de casos detectados con alguna variante estructural de hemoglobina fue 11 (2,17%) (Tabla 1), de los cuales 10 (1,97%) se clasificaron como heterocigotos para la HbS (rasgo falciforme) con fenotipo neonatal FAS y un caso (0.2%) fue portador de HbC con fenotipo neonatal FAC (Tabla 2). Todos los casos de hemoglobina S y hemoglobina C identificados por electroforesis se confirmaron por HPLC de fase reversa.
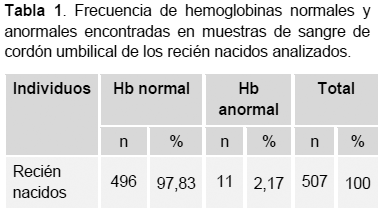

La figura 1 corresponde a una corrida electroforética en acetato de celulosa a pH 8.6 en la cual se observa una banda de Hb S que pertenece al paciente 458 (posición 3) del presente estudio. La figura 2 representa el cromatograma del mismo paciente: recién nacido con fenotipo neonatal FAS determinado por HPLC de fase reversa.

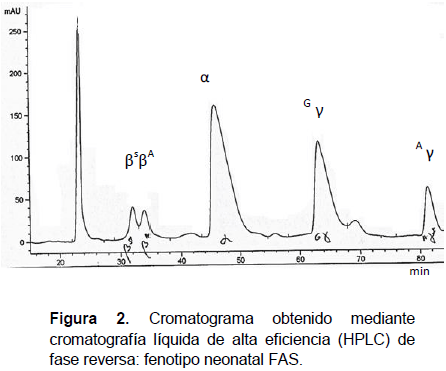
Por este método también se comprobó que ninguna muestra presentaba cadenas alfa o cadenas gamma (Hb F) anormales.
DISCUSIÓN
La distribución de las variantes de hemoglobina o hemoglobinas anormales varía de acuerdo con la población estudiada. Hasta el presente se han descrito más de 1.200 variantes de hemoglobina (13,14); éstas frecuentemente resultan de mutaciones puntuales en el gen de la globina ocasionando la sustitución de un aminoácido en una de las cadenas de la globina. Aunque muchas tienen poca importancia clínica, los homocigotos para HbS (anemia falciforme) y HbC (HbCC) presentan manifestaciones clínicas importantes así como los dobles heterocigotos SC. La HbS es la variante de mayor frecuencia a nivel mundial, aproximadamente 15% de los niños con anemia falciforme mueren debido a infecciones bacterianas graves, secuestro esplénico o crisis aplásicas u otras complicaciones propias de la enfermedad (2). En el continente americano la presencia de hemoglobinas anormales es producto de la inmigración y entrecruzamiento de diversas poblaciones africanas y europeas con los grupos amerindios, provocando una mezcla racial con la introducción de mutaciones en el gen de la globina. La población venezolana se ha originado principalmente de tres grandes ramas: indios (Caribes, Arawuacos), caucásicos (principalmente españoles e italianos) y africanos; las variantes hemoglobínicas S y C se introdujeron en el país con la llegada de los africanos en el siglo XVI (15) y su distribución pareciera seguir un patrón muy relacionado con el desplazamiento que las poblaciones de origen africano han tenido en el territorio nacional; la mayor frecuencia se encuentra en poblaciones costeras encontrán-dose una abrupta disminución en las poblaciones localizadas en la cordillera de los Andes (16).
Los resultados de este estudio demuestran que la HbS es la hemoglobinopatía estructural más frecuente en los recién nacidos estudiados en la ciudad de Valencia, siguiéndole en frecuencia la HbC, lo cual está de acuerdo con los resultados obtenidos por García y col. (17) en recién nacidos y con los estudios poblacionales realizados en adultos en varias regiones de Venezuela (4-6). Diferentes estudios realizados en el país confirman que las hemoglobinopatías son un problema de salud pública en Venezuela (4,5). La HbS es también la hemoglobinopatía de mayor prevalencia en recién nacidos de países como Colombia (18), Brasil (19,22), Cuba (23), Argentina (24). En este estudio no se detectaron individuos homocigotos para HbS ni para HbC, probablemente debido al tamaño de la muestra.
En la literatura existen diversas publicaciones relacionadas con la detección neonatal de hemoglobinopatías, lo cual demuestra la impor-tancia de este procedimiento en la reducción de la mortalidad en menores de 5 años (23,25-27). El diagnóstico neonatal en los países desarrollados junto con la profilaxis antibiótica ha reducido la mortalidad en los primeros años a casi cero (9,28). La alta frecuencia para HbS encontrada en los recién nacidos estudiados evidencia la necesidad de establecer un programa de detección neonatal de hemoglobinopatías.
Esto tiene dos ventajas: por un lado evitar las muertes prematuras en los neonatos enfermos, ya que permite instaurar un tratamiento profiláctico de antibióticos y otros cuidados especializados, evitando así la mortalidad por infecciones en los primeros años de vida. Si el programa incluye además educación a los padres y un seguimiento específico, se reduce sensiblemente la morbilidad y la mortalidad de la enfermedad en la lactancia y la primera infancia. Por otro lado, permite detectar portadores de un gen alterado, cuya descendencia podría presentar la enfermedad. Hay que tener presente que en la última década en Venezuela, aun cuando no existen datos oficiales, se ha producido un aumento del flujo de inmigrantes procedentes de China, lo que puede ocasionar cambios en el patrón de algunas enfermedades por la introducción de variantes propias a esa raza, por lo que debemos estar informados para detectarlas y tratarlas de modo adecuado. La Organización Mundial de la Salud ha recomendado que cada país realice programas particulares de detección de hemoglobinopatías de acuerdo con la incidencia local de la enfermedad, la estructura de los sistemas de salud y los recursos económicos (3,9).
Si bien los métodos tradicionales de electroforesis para la detección de hemoglobinopatías han ido sustituyéndose por HPLC de intercambio catiónico (CE), los métodos electroforéticos empleados en este estudio demostraron ser bastante sensibles para la detección de variantes estructurales en sangre de cordón, como ha sido demostrado en otros estudios (19,29,30); es por ello que se recomienda como una técnica válida para el tamizaje neonatal de las hemoglobinopatías, ya que reúne las características necesarias de una prueba adecuada para tal fin como son: rapidez, simplicidad, accesibilidad, fiabilidad y bajo costo.
Sin embargo, independientemente del método elegido para el despistaje inicial, éste no debe utilizarse como única prueba de detección, ya que en ocasiones, puede dar lugar a una identificación errada del fenotipo, requiriéndose la realización de métodos alternativos de confirmación.
En el presente estudio, las 11 muestras que arrojaron resultados anómalos por electroforesis fueron todas confirmadas por HPLC de fase reversa. Adicionalmente, se sugiere que los casos positivos sean estudiados nuevamente junto con su grupo familiar.
Esta investigación demuestra la importancia de plantear la creación de un programa regional para el tamizaje neonatal de hemoglobinopatías que, junto con un programa de educación y asesoría genética, prevendría la aparición de nuevos casos, y permitiría, además, el tratamiento precoz de los individuos homocigotos, lo cual favorecería la sobrevida del paciente y mejoraría significativamente su calidad de vida.
REFERENCIAS
1. García-Conde J, San Miguel J, Sierra J, Urbano-Ispizua A, Vicente V, Vives J. Hematología. Arán Ediciones, España 2003; pp 255-269. [ Links ]
2. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010; 115(22):4331-4336. [ Links ]
3. Organización Mundial de la Salud: Drepanocitosis y otras hemoglobinopatías. Nota descriptiva No.308, enero 2011. [ Links ]
4. Arends A, Chacín M, Bravo-Urquiola M, Montilla S, Guevara J, Velásquez D, García G, Álvarez M, Castillo O. Hemoglobinopatías en Venezuela. Interciencia 2007; 32(8):516-521. [ Links ]
5. Arends T. Epidemiología de las variantes hemoglobínicas en Venezuela. Gaceta Med. 1984; 92:189-224. [ Links ]
6. Arends T, Salazar R, Anchustegui M, Garlin G. Hemoglobin variant in the northeastern region of Venezuela. Interciencia 1990; 15:36-41. [ Links ]
7. Frenette P, Atweh G. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest 2007; 117(4):850-858. [ Links ]
8. Nagel R, Fabry M, Steinberg M. The paradox of hemoglobin SC disease. Blood Rev. 2003; 17:167-178. [ Links ]
9. Modell B, Darlison M. Global epidemiology of hemoglobin disorders and derived service indicators. Bull World Health Organ 2008; 86:480-487. [ Links ]
10. De Abajo F. La declaración de Helsinki VI: una revisión necesaria, pero ¿suficiente?. Rev Esp Salud Public 2001; 75(5):407-420. [ Links ]
11. Clarke G, Higgins T. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem 2000; 46 (8 Pt 2):1284-90. [ Links ]
12. Shelton JB, Shelton JR, Shroeder W. High performance liquid chromatographic separation of globin chains on a large-pore C4 column. J Liq Chromat 1984; 7(10):1969-1977. [ Links ]
13. Giardine B, van Beal S, Kaimakis P. Hb Var database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum Mut 2007; 28(2):206. [ Links ]
14. Globin Gene Server Hb Var a database of human hemoglobin variants and thalassemia Disp: http://globin.bx.psu.edu/hbvar/menu.html [ Links ]
15. Acosta-Saignes M. Vida de los esclavos negros en Venezuela. Hesperides. Caracas-Venezuela 1967; 412 pp. [ Links ]
16. Salazar-Lugo R. La hemoglobina S en la población venezolana. Invest Clin 2004; 45(2):175-183. [ Links ]
17. García O, Chacín M, Bravo M, Gómez G, Montilla S, Merzón R, Donato M, Castillo O, Arends A. Diagnóstico de hemoglobinopatías a partir de sangre del talón de recién nacidos en diferentes centros hospitalarios de Venezuela. An Pediatr 2009; 71(4):314-318. [ Links ]
18. De Bernal M, Collazos A, Bonilla R, Tascón E. Determinación de la prevalencia de la hemoglobina S, C, D y G en recién nacidos de Buenaventura, Colombia. Colomb Med 2010; 41(2):141-147. [ Links ]
19. Martini G, Meneghetti B, Santos N, Oliveira C, Ruhland L, da Silva P, Haas P. Triagem neonatal e hemoglobinopatías em Santa Catarina, Brasil. RBAC 2009; 41(3):185-189. [ Links ]
20. Campos L, Dias F, Mendes M. Hemoglobinas anormais em sangue de cordão umbilical. Rev. Bras. Hematol. Hemoter. 2006; 28(1):65-70. [ Links ]
21. Sommer C, Goldbeck A, Wagner S, Castro S. Triagem neonatal para hemoglobinopatias: experiência de um ano na rede de saúde pública do Rio Grande do Sul, Brasil. Cad. Saúde Pública 2006; 22(8):1709-1714. [ Links ]
22. Adorno E, Couto F, de Moura J, Figueiredo J, Rêgo M, Galvão dos Reis M, Souza M. Hemoglobinopatias em recém-nascidos de Salvador, Bahia, Nordeste do Brasil. Cad. Saúde Pública 2005; 21(1):292-298. [ Links ]
23. Taboada N, Gómez M, Algora A, Noa M, Arcas G, Noche G, Herrera M. Resultados del programa de prevención de hemoglobinopatías SS y SC en el periodo 1987-2007 en la provincia Villa Clara, Cuba. Rev Cubana Genet Comunit 2010; 4(1):37-41. [ Links ]
24. Noguera N, Bragós I, Morisoli L, Milani A. Screening for hemoglobinopathies in neonates in Argentina. Haematologica 1999; 84(5):468-470. [ Links ]
25. Gulbis B, Tshilolo L, Cotton F, Lin C, Vertongen F. Newborn screening for hemoglobinopathies: The Brussels experience. J Med Screen. 1999;6:11-5. [ Links ]
26. Githens JH, Lane PA, McCurdy RS, Houston ML, McKinna JD, Cole DM. Newborn screening for hemoglobinopathies in Colorado. The first 10 years. Am J Dis Child 1990;144:466-470. [ Links ]
27. Almeida AM, Henthorn JS, Davies SC. Neonatal screening for hemoglobinopathies: the results of a 10-year program in a English region. Br J Haematolo 2001; 112:32-5. [ Links ]
28. Angastiniotis M, Modell B, Englezos P, Boulyjenkov V. Prevention and control of hemoglobinopathies. Bull World Health Org 1995; 73:375-386. [ Links ]
29. Pignataro MC, Santos É, Pereira WA, Dantas TM. Prevalência de hemoglobinas anormais em recém-nascidos da cidade de Natal, Rio Grande do Norte, Brasil. Revista Cad. Saúde Pública, Rio de Janeiro 2004; 20(1):123-128. [ Links ]
30. Ducatti R, Teixeira A, Galão H, Bonini-Domingos C, Fett-Conte A. Investigação de hemoglobinopatias em sangue de cordão umbilical de recém-nascidos do Hospital de Base de São José do Rio Preto. Ver Bras Hematol Hemoter 2001; 23(1). [ Links ]
