










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. v.7 n.1 Mérida feb. 2009
Panhipopituitarismo secundario a macroadenoma hipofisario no funcionante en la adolescencia.
Yajaira Briceño, Jorge Chirinos, Mariela Paoli, Yajaira Zerpa
Unidad de Endocrinología, Instituto Autónomo Hospital Universitario de Los Andes, Mérida, Venezuela.
Dirigir correspondencia a: Yajaira Briceño. jmendoya@hotmail.com
Resumen
Objetivo: Presentar el caso poco frecuente de una adolescente con un macroadenoma hipofisario no funcionante que le produjo un hipopituitarismo. Se hace una revisión de la literatura.
Caso Clínico: Adolescente femenina de 16 años 2 meses de edad quien presenta poco progreso en talla y ausencia de caracteres sexuales secundarios; refiere concomitantemente cefaleas ocasionales. Al examen físico presenta talla y peso por debajo del pc 3, velocidad de crecimiento 2,8 cm/año, cubitus valgus, paladar ojival, cuarto metacarpiano corto, no tiromegalia. Genitales, mamas, vello axilar y púbico Tanner I. Se hace diagnóstico de talla baja patológica y retraso puberal y se indican exámenes de paraclínicos. Los resultados de laboratorio muestran un déficit de hormona de crecimiento (GH) y un hipocortisolismo, con función tiroidea conservada. Edad ósea de 11 años para una edad cronológica de 15 años. Cariotipo 46,XX. Rx lateral de cráneo muestra silla turca amplia y excavada. Us pélvico: útero en anteversion, central, con longitud de 20 mm; no se visualizan línea endometrial ni ovarios. La RMN muestra el piso de la región selar deformado, excavado, con imagen compatible con LOE de contornos más o menos definidos con señal intermedia baja y dishomogénea en T1, con realce hipertenso dishomogéneo tras la administración del medio de contraste, de aproximadamente 2,9 x 1,36 cm. de diámetro, con compromiso de los elementos supraselares, ejerciendo efecto compresivo sobre el infundíbulo el cual deforma el quiasma óptico. Se hace diagnóstico de Panhipopituitarismo secundario a Macroadenoma Hipofisario no Secretante, con Déficit de GH, Hipogonadismo Hipogonadotropo e Insuficiencia Suprarrenal Secundaria (Déficit de ACTH). Se indica tratamiento con hidrocortisona por vía oral. Se refiere para resolución quirúrgica por vía transesfenoidal ya que en nuestro centro no se realiza. Se desconoce el reporte histológico del tumor y la evolución de la paciente.
Conclusiones: Los adenomas hipofisarios representan menos del 2-3% de todos los tumores intracraneales y sus manifestaciones clínicas dependen de la suma de un efecto masa, que causa alteraciones neurológicas, y la afectación de la secreción hormonal, ya sea por exceso o por defecto. Es importante realizar un diagnóstico temprano, así como un tratamiento efectivo y seguimiento a largo plazo.
Palabras clave: Tumores Hipofisarios, Macroadenoma Hipofisario, Panhipopituitarismo.
Abstract
Objective: To present the uncommon case of a teenager with a non-functioning pituitary macroadenoma that resulted in a hypopituitarism. A literature review is done.
Case Report: Female adolescent of 16 years 2 months of age who presents little progress in stature and absence of secondary sexual characters; concomitantly refers occasional headaches. Physical examination: height and weight below the 3 pc, growth velocity 2.8 cm/year, cubitus valgus, ojival palate, short fourth metacarpal, no thyroid enlargement. Genitals, breasts, axillary and pubic hair, Tanner I. The diagnosis of pathological short stature and delayed puberty is done and paraclinical examinations are indicated. Laboratory results show a deficit of growth hormone (GH) and a hypocortisolism with preserved thyroid function. Bone age of 11 years for a chronological age of 15 years. Karyotype 46, XX. Rx of the skull shows a wide and excavated sella turcica. Pelvic Us: central uterus, in anteversion, 20 mm in length and the endometrium and ovaries were no visible. NMR shows the floor of the sellar region warped, carved, with a image compatible with tumour of defined contours, with low-intermediate and dishomogenea signal on T1, with hypertensive and heterogeneou enhancement after administration of contrast medium, approximately 2.9 x 1.36 cm. in diameter, with involvement of the supraselares elements exerting compressive effect on the infundibulum, which deforms the optic chiasm. The diagnosis of panhypopituitarism secondary to pituitary non functioning macroadenoma is done, with GH deficiency, hypogonadotropic hypogonadism and secondary adrenal insufficiency (ACTH deficiency). Treatment with oral hydrocortisone is indicated. The patient is referred for resolution by transsphenoidal surgery. The histological report of the tumor and the evolution of the patient are unknown.
Conclusions: Pituitary adenomas account for less than 2-3% of all intracranial tumours, and their clinical manifestations depend on the sum of a mass effect, which causes neurological disorders, and the disruption of hormonal secretion, either up or down. It is important to make an early diagnosis, an effective treatment and long-term monitoring.
Key words: Pituitary tumours, pituitary macroadenoma, panhypopituitarism.
Artículo recibido en: Noviembre 2008. Aceptado para publicación en: Diciembre 2008.
Introducción
La mayoría de los tumores hipofisarios son benignos, de crecimiento lento y se originan de uno de los cinco tipos celulares de la hipófisis anterior. Son raros en niños y adolescentes, representan menos del 2-3% de todos los tumores intracraneales1-4. No se reporta en la literatura la prevalencia de macroadenomas hipofisarios en la adolescencia.
El hipopituitarismo puede presentarse con una deficiencia hormonal aislada o con deficiencia de múltiples hormonas (panhipopituitarismo).
La etiología es variada, abarca desde deficiencias primarias a las secundarias tales como los tumores hipofisarios o supraselares, lesiones infiltrativas o destructivas que siguen a la lesión cerebral o a la radioterapia. Las manifestaciones clínicas se deben al efecto local de ocupación de espacio y a los síndromes de hiposecreción o hipersecreción hormonal producidos directamente por el adenoma o surgidos como consecuencia del tratamiento.
Estos pacientes necesitan tratamiento y seguimiento durante el resto de su vida. Los tumores hipofisarios se clasifican de acuerdo al tamaño, así menores de 10 mm (1 cm.) corresponde a los microadenomas y mayores de 1 cm. a los macroadenomas, esto tiene valor pronóstico, puesto que los tumores más grandes suelen tener más dificultades a la hora del tratamiento y un mayor índice de recidivas posterapias. La clasificación más usada por la O.M.S. tiene en cuenta cinco características de los adenomas: actividad endocrina, imagen radiológica e intraoperatoria, histología, inmunohistoquímica y ultraestructura2,3.
Los tumores hipofisarios pueden presentarse clínicamente como un cuadro de hiperfunción, condicionado por un exceso de secreción de una hormona determinada, o por un cuadro de hipofunción, generalmente debido a la destrucción glandular por la compresión que provoca el crecimiento tumoral. La hipofunción hormonal también puede ser motivada por el efecto fisiológico inhibidor que algunas hormonas tienen sobre la secreción de otras2-4.
Caso Clínico
Se trata de adolescente femenina de 16 años 2 meses de edad quien inicia enfermedad actual a los 8 años de edad caracterizada por poco progreso en talla y ausencia de caracteres sexuales secundarios: refiere concomitantemente cefaleas ocasionales. Antecedentes de importancia: Madre sana, Menarquia 13 años, Talla 156 cms. Padre sano, Talla 160 cms. Potencial Genético: 156,5 ± 9 cms. Al examen físico de ingreso se reporta Talla 129,9 cm. (por debajo del pc 3), edad talla de 8,9 años, peso de 29,4 kg. (por debajo del pc 3) con peso ideal para la talla de 28 kg. Velocidad de crecimiento entre mayo de 2006 y mayo de 2007 de 2,8 cm/ año (por debajo del pc 3). Brazada 127 cms, segmento superior 62 cms, segmento inferior 65 cms, relación segmento sup/inf: 0,9. Se observa cubitus valgus, paladar ojival, cuarto metacarpiano corto. No tiromegalia. Genitales: Mamas y vello axilar y púbico Tanner I. Se hace diagnóstico de talla baja patológica y retraso puberal y se indican exámenes paraclínicos.
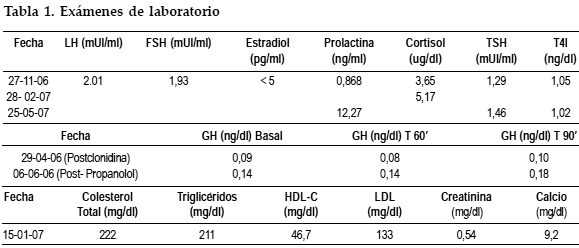
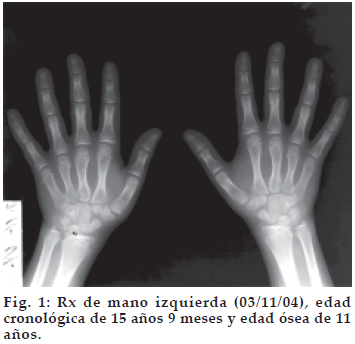
Los resultados de laboratorio muestran un déficit de hormona de crecimiento (GH) y un hipocortisolismo, con función tiroidea conservada (Tabla I). La edad ósea según el atlas de maduración ósea del venezolano fue de 11 años, para una edad cronológica de 15 años (Figura 1). El cariotipo en sangre periférica 30 metafases reporta 46 XX. Rayos X lateral de cráneo demuestra una silla turca amplia y excavada (Figura 2). El ultrasonido pélvico de fecha 18-05-05, muestra útero en AV, central, con longitud de 20 mm; no se visualizan línea endometrial ni ovarios. La Resonancia Magnética Cerebral (19-10-06) demuestra el piso de la región selar deformado, excavado, con imagen compatible con LOE de contornos más o menos definidos con señal intermedia baja y dishomogénea en T1, con realce hipertenso dishomogéneo tras la administración del medio de contraste, de aproximadamente 2,9 x 1,36 cm. de diámetro, con compromiso de los elementos supraselares, ejerciendo efecto compresivo sobre el infundíbulo el cual deforma el quiasma óptico (Figura 3). Se hace diagnóstico de Panhipopituitarismo secundario a Macroadenoma Hipofisario no Secretante, con Déficit de GH, Hipogonadismo Hipogonadotropo e Insuficiencia Suprarrenal Secundaria (Déficit de ACTH). Se indicó tratamiento con Hidrocortisona por vía oral. En Mayo del 2007 se decide referencia de la paciente al Hospital Universitario de Caracas para resolución quirúrgica por vía transesfenoidal ya que en nuestro centro no se cuenta con los equipos necesarios para la realización de dicha cirugía.


La paciente no ha asistido más a consulta posterior a la cirugía por lo que se desconoce el reporte histológico del tumor y la evolución de la paciente.
Discusión
Los adenomas hipofisarios son generalmente neoplasias benignas originadas en uno de los cinco tipos celulares de la hipófisis anterior y son de crecimiento lento. El fenotipo clínico y bioquímico de estos tumores depende del tipo celular del que proceden, pueden originarse a partir de un solo tipo celular o estar formados por células con funciones distintas dentro del mismo tumor. Los tumores con actividad hormonal se caracterizan por tener secreción autónoma, con escasa repuesta a las vías fisiológicas normales de inhibición1.
El hipopituitarismo puede presentarse como una deficiencia hormonal aislada o como de múltiples hormonas (panhipopituitarismo), este último fue el cuadro clínico que se presentó el caso en estudio. La etiología es variada y abarca desde deficiencias primarias y secundarias; en los hipopituitarismos primarios se mencionan la ausencia o destrucción de las células hipofisarias por tumores hipofisarios (lo más frecuente) y otras causas (aneurismas, necrosis isquémica de la hipófisis, traumatismos craneales, infecciones, granulomas, autoinmune, hemocromatosis, metástasis, iatrógenos por cirugía o radioterapia); en los secundarios se describen alteraciones del tallo hipofisario (traumatismos y cirugía, tumores extrahipofisarios o extraselares y aneurismas) o alteraciones hipotalámicas (tumores, granulomas, traumatismos, malnutrición, anorexia nerviosa) o causas iatrogénicas (tratamientos prolongados con glucocorticoides, anticonceptivos, citostáticos o irradiación)2.
Los adenomas hipofisarios constituyen una patología poco frecuente, con una incidencia anual de 0,1/100.000 niños. En la edad pediátrica, representan menos del 2-3% de todos los tumores intracraneales, menos del 3% de los tumores supratentoriales y el 2 a 6% de los adenomas hipofisarios intervenidos3. Más de dos tercios de los tumores se presentan en la pubertad, quizás por ser éste un período caracterizado por la aceleración del crecimiento en todos los tejidos del organismo, lo que hace más susceptibles a las células hipofisarias de sufrir los cambios que inician la génesis tumoral4.
Actualmente se acepta como mecanismo etiopatogénico de la génesis tumoral la presencia de dos estadíos diferentes, inicio y promoción. En un primer momento, las células hipofisarias normales sufrirían mutaciones, espontáneas y adquiridas, que llevarían a la expresión de anomalías celulares, y en un segundo momento las hormonas hipotalámicas y los factores de crecimiento actuarían como factores permisivos, potenciando el desarrollo de un clon celular, y con ello, la producción de uno o más tipos hormonales5.
El hipopituitarismo puede presentarse en adenomas hipofisarios secretores y no secretores. Los macroadenomas invaden estructuras vecinas; si la silla turca está intacta son de clase II, cuando producen ensanchamiento de la silla turca son de clase III y los que presentan invasión y destrucción son clase IV. Los macroadenomas tienen tendencia a producir síntomas por su tamaño. Ambos micro y macroadenomas pueden ser activos hormonalmente. Los tumores hipofisarios también son clasificados como funcionales (60%), es decir que producen en exceso una o más hormonas, o no funcionales (40%), que no producen hormonas y constituyen solo masas de células. Los funcionantes se clasifican y nombran según la hormona que producen, prolactinomas (prolactina) que son el subtipo mas frecuente, seguidos por los somatotropos o productores de GH y corticotropos o productores de ACTH (Enfermedad de Cushing)6,7. El caso que se presenta se trata de una macroadenoma no funcionante.
Las manifestaciones clínicas dependen de la suma de un efecto masa, que causa alteraciones neurológicas, predominando en los tumores hipotalámicos y en los macroadenomas, y la afectación de la secreción hormonal, ya sea por exceso o por defecto. Los tumores hipofisarios pueden presentarse clínicamente como un cuadro de hiperfunción, condicionado por un exceso de secreción de una hormona determinada, o por un cuadro de hipofunción, generalmente debido a la destrucción glandular por la compresión que provoca el crecimiento tumoral (hipopituitarismo)8. Los síntomas de hipofunción más frecuentes son el retraso del crecimiento, por el déficit de GH y la afectación del desarrollo puberal por el hipogonadismo. En el caso que se presenta, es evidente una detención del crecimiento y talla baja, además de retraso puberal completo, sin caracteres sexuales secundarios a los 16 años de edad. Las determinaciones hormonales demostraron deficiencias múltiples, de GH, de gonadotropinas y de cortisol, con preservación de la función tiroidea. Además, aunque esta paciente solo refería cefalea, en los macroadenomas es posible que el compromiso de las estructuras vecinas provoque síntomas característicos como la diabetes insípida. La vecindad del quiasma óptico determina su posible compresión por la extensión suprasellar del tumor. La presión selectiva sobre un nervio óptico podría provocar pérdida de visión del ojo afectado y la compresión sobre el quiasma podría causar cuadrantanopsia bitemporal, que si sigue progresando, puede transformarse en hemianopsia bitemporal, por esta razón la campimetria, ya sea por confrontación o asistida, es una prueba necesaria, teniendo como inconveniente la necesidad de colaboración por parte del paciente. La expansión lateral del adenoma puede causar afectación de los pares craneales III, IV y VI, cursando con ptosis palpebral, midriasis, oftalmoplejia y diplopía. También se ha descrito la presencia de hidrocefalia si se bloquea el agujero de Monroe y de licuorrea si se perfora el suelo de la silla turca; un cuadro más raro en niños es la apoplejía hipofisaria, causado por una expansión aguda del adenoma por un infarto hemorrágico intratumoral9,10.
El diagnóstico del adenoma se hace a través de pruebas bioquímicas, de modo que mediante la determinación de los niveles basales de las hormonas se puede establecer una situación de hipofunción o de hiperfunción. El desarrollo de la radiología intervencionista permite tomar muestras hormonales de la sangre venosa que abandona cada hemihipófisis, mediante la colocación de catéteres en el seno petroso inferior, con el fin de localizar con más exactitud un microadenoma. El diagnóstico también se realiza mediante pruebas de imagen; la radiografía lateral de cráneo todavía tiene alguna vigencia en tumores grandes, aunque actualmente la TAC y de preferencia la RMN son las técnicas más sensibles, ya que permiten obtener imágenes claras y directas de la hipófisis, del hipotálamo y de las regiones vecinas. Su eficacia llega a la detección de lesiones de 3-4 mm, con cifras de sensibilidad superiores al 90%, sobre todo al realzar las imágenes con gadolinio. Se están desarrollando nuevas técnicas para completar la eficacia diagnóstica, como la ecografía intraoperatoria, la tomografía de emisión de positrones (PET) que utiliza como trazador 18F-desoxiglucosa, un elemento que es captado, casi en exclusiva, por el tejido tumoral; por último el octreoscan, modalidad de gammagrafía que utiliza como trazador un análogo de la somastotastina marcado, aprovecha la propiedad de expresar receptores de somastotastina que tienen algunas células tumorales para aportar una imagen morfofuncional11. En nuestra paciente fue evidente la presencia de la tumoración con la Rx lateral de cráneo, donde se demostró una silla turca amplia y excavada (grado III); esto es infrecuente de hallar, principalmente porque la mayoría de los adenomas hipofisarios en niños y adolescentes, que de por sí son raros, son pequeños, son microadenomas, los cuales no son capaces de alterar la morfología de la silla turca.
El diagnóstico de adenoma obliga a un tratamiento dirigido a eliminar la masa tumoral, preservar el tejido glandular sano, corregir el funcionamiento y prevenir las recurrencias; para ello se cuenta con tres técnicas: cirugía, farmacología y radioterapia. Estos pacientes necesitan tratamiento y seguimiento durante el resto de su vida. Los nuevos adelantos de la cirugía transesfenoidal que se realiza en más del 95% de los casos y los agentes terapéuticos más modernos, han mejorado el tratamiento de los tumores hipofisarios. La cirugía es eficaz en el 70-91% de los microadenomas, en el caso de los macroadenomas las cifras bajan a un 40- 56% de remisiones10. Un estudio reciente refiere que la función hormonal puede mejorar en el 50% de los pacientes después de la adenomectomía transesfenoidal12. Se ha realizado también abordaje endoscópico transesfenoidal con buenos resultados y la ventaja de ser un método menos invasivo y con menores complicaciones13.La radioterapia estereotáctica (incluida la radioterapia con bisturí gamma) constituye en la actualidad una segunda línea terapéutica en aquellos pacientes que sufren una recidiva o en los que la evaluación postquirúrgica revela la existencia de restos tumorales. El objetivo de dicho tratamiento es la normalización del exceso de secreción hipofisaria, la mejoría de los síntomas y signos de los síndromes de hipersecreción hormonal y la reducción o ablación de las grandes masa tumorales con el consiguiente alivio de la compresión de las estructuras adyacentes14,15. Lamentablemente no se ha podido realizar el seguimiento apropiado de la paciente.
Conclusión
Se presenta el caso poco frecuente de una adolescente con talla baja y retraso puberal debido a la presencia de un macroadenoma hipofisario no secretante, con múltiples déficits hormonales corroborados. Se indicó tratamiento médico para suplir el déficit de ACTH, y tratamiento quirúrgico (cirugía transesfenoidal) para extirpación del tumor. De acuerdo al reporte histológico e inmunohistoquímico del tumor, se debe planificar el tratamiento médico para la corrección del déficit de GH con miras a mejorar su talla final, y del hipogonadismo. Se debe realizar siguiendo los lineamientos para el uso de GH en casos de tumoraciones. Igualmente, es necesario el seguimiento a largo plazo tanto del hipopituitarismo como de una posible recidiva de la lesión tumoral.
Referecias Bibliográficas
1. Pérez-Higueras H. Exploración Neuroradiológica de la región hipotálamo-hipofisaria. En: Argente A, Carrascosa A, Gracia R, Rodríguez F. Tratado de Endocrinología Pediátrica y de la Adolescencia. Segunda Edición. Ediciones Doyma. Barcelona. 2000: 599-619. [ Links ]
2. Aron D, Finding J, Tyrrel J. Hipotálamo e hipófisis. En: Greenspan F, Strewler G. Endocrinología básica y clínica. Editorial Manual Moderno Cuarta Edición. México 2000: 107-179. [ Links ]
3. Melmed S, Larry J. Trastornos de la adenohipófisis y del hipotálamo. En: Larry J. Harrisons Endocrinology. Primera Edición. Editorial McGraw-Hill, USA 2006:19-56. [ Links ]
4. Fernández J, Casanueva F. Tumores Hipofisarios. En: Pombo, Tratado de Endocrinología Pediátrica. Tercera Edición. Editorial McGraw-Hill. Interamericana, España 2002:516-528. [ Links ]
5. Rosenfeld R, Cohen P. Disorders of growth hormone/insulin like growth factor secretion and action. In: Sperling M. Pediatric Endocrinology. Second Edition. Saunders Editorial, USA 2002: 211-288. [ Links ]
6. Money J. Hormones, hormonal anomalies, and psychological health care. In: Wilkins. The Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence. Fourth Edition. Charles C Thomas. Publisher. USA 1994:1141- 1178. [ Links ]
7. Nuñez de la Vega JM, Ramos R. Patogénesis de los adenomas hipofisiarios. Reporte de un caso y revisión de la literatura. Revista Mexicana Neurocirugía. 2006; 7:69–71. [ Links ]
8. Asa Sl, Ezzat S. The pathogenesis of pituitary tumor. Nature Neuroscience 2002; 2: 836- 49. [ Links ]
9. Escobar A. Tumores de la hipófisis. Revista Mexicana Neurocirugía. 2006; 7: 586–591. [ Links ]
10. Naoko S, Akira T, Yoshiyuki O, Eva H, Kalman K, Lloyd R, Scheithauer B. Pathology of pituitary tumors. Neurosurgery Clinics of North America 2003; 14: 25–39 [ Links ]
11. Neoplasias Intracraneales y trastornos paraneoplásicos. En: Raymond A, Maurice V, Allan R. Principios de Neurología. Séptima Edición McGraw-Hill México.2006:559-604. [ Links ]
12. Fatemi N, Dusick JR, Mattozo C, McArthur DL, Cohan P, Boscardin J, Wang C, Swerdloff RS, Nelly DF. Pituitary hormonal loss and recovery alter transsphenoidal adenoma renoval. Neurosurgery 2008; 63: 709-718. [ Links ]
13. Rudnik A, Zawadzki T, Wojtacha M, Bazowski P, Zubgaluszka-Ignasiak B, Duda I. Endoscopio transsesphenoidal treatment of pituitary adenomas. Neurol Neurochir Pol 2005; 39: 17-23. [ Links ]
14. Depper M, Hart L. Tumores encefálicos pediátricos. En: Orrison W Jr. Neuroradiología. 1ª Edición. Madrid-España. 2001:1550-1542. [ Links ]
15. Navarro C, Uribe C. Tumores del Sistema Nervioso Central. Sindrome de Hipertensión Endocraneana. En: Vélez H, Rojas W, Borrero J, Restrepo J. Fundamentos de Medicina. Neurología. 5ª Edición. Medellín Colombia.1996: 420-455. [ Links ]

