










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. v.7 n.3 Mérida sep. 2009
Síndrome de Cutis Marmorata Telangiectásica Congénita o Síndrome de Rothmund-Thompson.
Miguel Sampedro
Servicio de Endocrinología, Hospital Metropolitano del Norte, Valencia, Venezuela
Dirigir correspondencia a: Dr. Miguel Sampedro. miguelsampedro@cantv.net
RESUMEN
Objetivos: Presentar el caso de un síndrome muy raro, el Síndrome de Rothmund-Thomson, de herencia autosómica recesiva, atribuible a una mutación en el gen RECQL4 helicase, 8q24. Se caracteriza por la presencia de placas cutáneas reticuladas, atróficas, hiperpigmentadas, telangiectásicas, que a menudo se acompañan de catarata juvenil, nariz en silla de montar, defectos óseos congénitos, trastornos en el crecimiento del cabello, uñas, dientes, hipotiroidismo, talla baja e hipogonadismo.
Caso clínico: Paciente femenina de 12 años que consulta por talla baja. Antecedentes personales y familiares sin importancia. La familia ha notado cambios cutáneos en la piel desde poco después del nacimiento. La niña tiene un retraso escolar importante. Al examen físico presenta talla 124 cm, peso 21 kg, ambos muy por debajo del percentil 3 para su edad y sexo. Facies de cara de pájaro, piel pálida con telangiectasias rojas en telaraña en cara, brazos y abdomen, alopecia difusa, paladar ojival, atrofia cutánea en manos y atrofia de uñas, dientes irregulares y malformados, incisivos proyectados hacia delante. Presenta bocio difuso grado Ib, telarquia estadío II de Tanner (botón mamario) y no hay pubarquia. Edad ósea de 10 años. Con los exámenes de laboratorio se diagnostica hipotiroidismo primario y se indica tratamiento con 50 μg de levotiroxina sódica. En el seguimiento a los 3 meses se nota crecimiento de 2 cms y normalización de TSH. Evaluación oftalmológica sin alteraciones. Dada la presentación clínica y la evolución de la paciente, se establece el diagnóstico de Síndrome de Rothmund Thomson.
Conclusiones: El síndrome de Rothmund Thompson es una entidad clínica poco frecuente, asociada con una amplia gama de alteraciones endocrinas, por lo cual consideramos importante reportar este caso.
Palabras clave: Síndrome de cutis marmorata o Rothmund-Thomson, Poiquiloderma congénita, Poiquiloderma atrófico y catarata.
ABSTRACT
Objectives: To present the case of a rare syndrome, the Rothmund-Thomson syndrome, autosomal recessive, attributable to a mutation in the RECQL4 helicase gene, 8q24. It is characterized by reticulate skin plaques, atrophic, hyperpigmented, telangiectatic, often accompanied by juvenile cataracts, saddle nose, congenital bone defects, disturbances in the growth of hair, nails, teeth, hypothyroidism, short stature and hypogonadism.
Clinical case: A twelve year old girl came to the clinic because of short stature. Her personal and family history was unremarkable. Her parents noticed skin changes few days after birth. She has a delay in her school performance. Physical exam: height: 124cm, weight: 21kg, both below the third percentile for her age and sex; a bird face appearance. Skin pale with red spider thelangiectasias in the face, arms and abdomen. Generalized alopecia. Arched palate. There was skin and nail atrophy in both hands, irregular and malformed teeth and incisors projected forward. Goiter grade Ib, breasts with budding tanner stage II, there was no pubic hair. Bone age of 10 years. Laboratory tests: a diagnosis of primary hypothyroidism was made. She was started on L-Thyroxine 50 micrograms daily with an increase of 2cm when she came back three months later. An ophthalmological evaluation was normal. With the above clinical findings and clinical course we conclude that she has the Rothmund-Thomson Syndrome.
Conclusions: The Rothmund Thompson syndrome is a rare clinical entity associated with a wide range of endocrine disruption, so we consider it is important to report this case.
Key words: Cutis marmorata syndrome or Rothmund-Thomson syndrome. Congenital poikilodermia. Atrophyc poikilodermia and cataracts.
Artículo recibido en: Marzo 2009. Aceptado para publicación en: Mayo 2009
INTRODUCCIÓN
El Síndrome de Rothmund-Thomson es una rara afección descrita por el oftalmólogo alemán Rothmund en 1868, y publicada por primera vez por Van Lohuizen1 en 1922. Tiene varios sinónimos: Poiquiloderma Congénito, Poiquiloderma Atrófico pero el más aceptado hoy en día es Cutis Marmorata Telangectásica Congénita. Desde el punto de vista cutáneo, se caracteriza por una marcada fotosensibilidad y lesiones de características poiquilodérmicas2-4. Este síndrome es una genodermatosis autosómica recesiva atribuible a mutaciones del gen RECQL4 en 8q24, el cual codifica una RecQ DNA helicase5-8. Su predominio en relación al sexo no está claro. En más del 90% de los pacientes la sintomatología comienza a desarrollarse entre el tercer y sexto mes de vida mediante la presencia de placas eritematosas, de aspecto reticulado, a veces con leve hiperqueratosis, que a lo largo de su evolución, dejan hipo e hiperpigmentaciones residuales, lo cual le confiere a la zona afectada su tan característico aspecto poiquilodérmico. Muy raras veces se presentan en el recién nacido. En la mayoría de los casos reflejados en la literatura médica las lesiones comienzan en la cara y se extienden a los glúteos y la superficie extensora de las extremidades. Además presenta una red vascular reticulada profunda, eritematosavioleta, localizada o generalizada presente al nacer. Entre las manifestaciones del área endocrina se encuentran: disfunción tiroidea, talla baja y retraso puberal2-4. Muy pocos casos han sido descritos en la literatura mundial, se totalizan unos 300 reportados, por lo que consideramos importante, reportar este caso.
CASO CLÍNICO
Se trata de una paciente femenina de 12 años que consulta por talla baja. Los antecedentes personales y familiares son sin importancia hasta la tercera generación, no existiendo antecedentes de lesiones cutáneas en la familia. La familia ha notado estos cambios cutáneos en la piel desde poco después del nacimiento pero los médicos que han consultado no llegaron a ninguna conclusión (no consultaron ni con Dermatólogo ni con Endocrinólogo). La niña tiene un retraso escolar importante y recibe clases particulares desde hace tiempo. Al examen físico presenta talla de 124 cm y peso de 21 kg, ambos muy por debajo del percentil 3 para su edad y sexo. Se observa piel pálida con telangiectasias rojas en telaraña en cara, brazos y abdomen, que se acentúan con el frío, emociones y llanto (Fotos I y II), alopecia difusa, paladar ojival, atrofia cutánea en manos y atrofia de uñas, dientes irregulares y malformados, incisivos proyectados hacia delante, cara de pájaro. Se palpa bocio difuso grado Ib y con respecto al desarrollo puberal, presenta telarquia en estadio II de Tanner (botón mamario) y no hay pubarquía. No hay dismorfismo característico excepto por la cara de pájaro (Foto III).
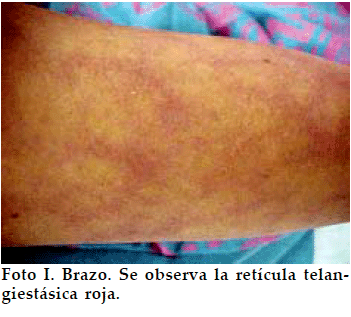
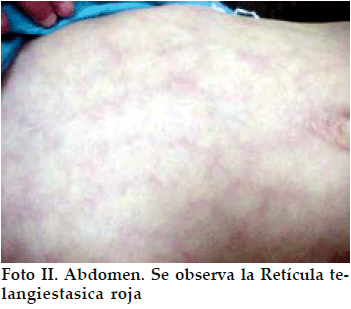
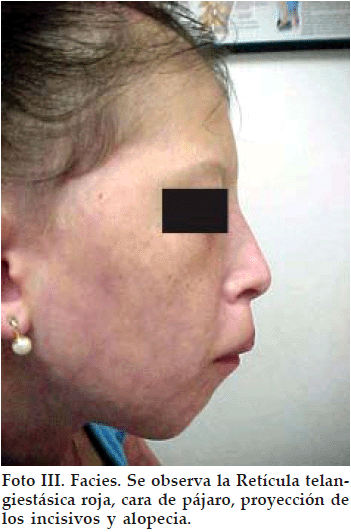
Se realizan Examenes complementarios que reportan: T4L 0.7 µg/dL (VN: 0.8 a 1.2); TSH 6 U/L (VN 0.5 a 4); hGH basal 22 µg /mL (VN 0- 10) y tras ejercicio 40 µg /mL; hGH basal 4.5 y tras Clonidina 17 µg /mL; Hb 11.9 g/dL; Proteinas totales 5.8 g/dL; Albumina 3.3 g /dL; Globulina 2.5 g/dL. La tomografia axial computarizada (TAC) cerebral fue normal. La edad ósea fue de 10 años.
Con estos resultados se hace diagnostico de hipotiroidismo primario y se indica tratamiento con 50 µg de levotiroxina sodica. En el seguimiento a los 3 meses se nota crecimiento de 2 cms y la TSH en 1.6 mU/mL, lo que sugiere una buena respuesta clinica al tratamiento. Las proteinas plasmaticas aumentaron a 6.9 g/dL. Actualmente la evaluacion oftalmologica no revela ningun tipo de lesion. Dada la presentacion clinica y la evolucion de la paciente, se establece el diagnostico de Sindrome de Rothmund Thomson.
DISCUSIÓN
El síndrome de Rothmund-Thomson es una patología muy rara que afecta varias partes del cuerpo, en especial la piel. Generalmente va asociado a hipotiroidismo congénito o infantil3. Estas alteraciones estaban presentes en nuestra paciente. Con el transcurrir del tiempo, las lesiones dermatológicas se extienden a brazos y piernas, causando cambios en la coloración de la piel, ulceraciones, áreas de degeneración tisular, atrofia cutánea e hipertrofia o atrofia del miembro afectado. Los problemas en piel persisten de por vida y son colectivamente denominados poiquilodermia1-3.
Se han reportado casos con cataratas congénitas o precoces y glaucoma-2-5. En nuestra paciente no se observaron alteraciones oftalmológicas. Generalmente el síndrome se acompaña de alteraciones esqueléticas y articulares, alteración en los dientes, talla baja, e inclusive enanismo. En la paciente se observaron alteraciones en los dientes y talla baja. En un 25% de los casos se ha reportado hipogonadismo; nuestra paciente ya inició pubertad a los 12 años, por lo que no parece haber retraso puberal. Los diferentes síntomas y signos descritos para el síndrome de Rothmund-Thomson se superponen con los de otras patologías, como son los síndromes de Baller- Gerold y Rapadilino, en los cuales también se presentan defectos radiales, anomalías esqueléticas y crecimiento lento. Estos síndromes tienen un denominador común y es la mutación en el mismo gen. Basados en estas similitudes, hoy día se plantea como interrogante determinar si estos síndromes son entidades clínicas diferentes o son parte de un solo síndrome cuyos síntomas y signos se sobreponen9. El mecanismo que subyace al desarrollo de la sintomatología cutánea en estos pacientes parece ser una alteración en los mecanismos de reparación del ADN (la alteración del gen que codifica la helicasa RECQL4, parece ser responsable de muchos de estos casos)3. Esto se traduce en una importante inestabilidad cromosómica en la síntesis del ADN en los fibroblastos, acentuada sobre todo tras la exposición a la radiación ultravioleta. En nuestra paciente y su familia no se ha podido realizar estudio genético por motivos técnicos, aunque consideramos interesante que se lleve a cabo, si bien los patrones genéticos del síndrome de Rothmund-Thomson son variados. Es interesante y será objeto de futuros estudios el establecer una correlación entre las alteraciones en el genotipo y su expresión fenotípica10.
Desde el punto de vista dermatológico es preciso estar atento al posible desarrollo de neoplasias cutáneas en edades más precoces que en el resto de la población10,11. También se han descrito fibrosarcomas, mielodisplasia, adenomas de paratiroides, carcinomas gástricos y osteosarcomas asociados11,12. Esto hace imprescindible insistir en las correctas medidas de fotoprotección que se deben aplicar.
REFERENCIAS BIBLIOGRAFICAS
1. Van Lohuizen CHJ. Uber eine seltene angeborene Hautanomalie (Cutis marmorate telangiestatica congénita). Acta Derm Venereol (Stockh) 1922; 3: 202-211. [ Links ]
2. Silverberg NB, Biro DE, Laude TA. What syndrome is this? Rothmund-Thomson syndrome (poikiloderma congenitale). Pediatr Dermatol 1999;16:59-61. [ Links ]
3. Picascia DD, Easterly NB. Cutis marmorata telangectasia congenita: A report of 22 Cases. J Am Acad Dermatol 1989; 20: 1098-1101. [ Links ]
4. Ruiz Villaverde R, Alonso Corral MJ, Sánchez Cano D, Pacheco Sánchez-Lafuente FJ. Sindrome de Rothmund-Thomson . An Pediatr (Barc) 2005;63:271-272. [ Links ]
5. Wu J.H., Capp C., Feng L.P., Hsieth T.S. Drosophila homologue of the Rothmund Thompson Syndrome Gene: Essential function in DNA replication during development. Devl Biol 2008; 323:130-142. [ Links ]
6. Siitonen, H.A. The mutation spectrum in RECQL4 diseases. Eur J Gen 2009;17: 151-158. [ Links ]
7. Xu X.H., Lui Y.L. Dual DNA unwinding activities of the Rothmund- Thomson syndrome Protein REQ4. EMBO J 2009;28: 568 -577. [ Links ]
8. Mehollin Ray A.R. Radiographic abnormalities in Rothmund- Thomson syndrome and genotype correlation with REQL4 mutation status. Am J Radiol 2008;191: W62-W66. [ Links ]
9. Piquero-Casals J, Okubo AY, Nico MM. Rothmund-thomson syndrome in three siblings and development of cutaneous squamous cell carcinoma. Pediatr Dermatol 2002;19:312-316. [ Links ]
10. Cabral RE, Queille S, Bodemer C, de Prost Y, Neto JB, Sarasin A, Daya-Grosjean L. Identification of new RECQL4 mutations in Caucasian Rothmund-Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat Res 2008;643:41-47. [ Links ]
11. Marin-Bertolin S, Amorrortu-Velayos J, Aliaga Boniche A. Squamous cell carcinoma of the tongue in a patient with Rothmund-Thomson syndrome. Br J Plast Surg 1998;51:646-648. [ Links ]
12. Pianigiani E, De Aloe G, Andreassi A, Rubegni P, Fimiani M. Rothmund-Thomson syndrome (Thomson-type) and myelodysplasia. Pediatr Dermatol 2001;18:422-425. [ Links ]

