










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Venezolana de Endocrinología y Metabolismo
versión impresa ISSN 1690-3110
Rev. Venez. Endocrinol. Metab. vol.13 no.2 Mérida jun. 2015
Feocromocitoma gigante abscedado: A propósito de un caso.
Jenny De Jesús1, Franklin García1, 2, Liliana Fung1, Evelyn Hernández1.
1 Servicio de Endocrinología y Enfermedades Metabólicas.
2 Cátedra de Clínica y Terapéutica Quirúrgica D, Servicio de Cirugía IV. Hospital Universitario de Caracas. Caracas, Venezuela.
Dirigir correspondencia a: Franklin, García MD. Email: garcifra1@gmail.com
RESUMEN
Objetivos: Describir la presentación de un caso clínico de feocromocitoma gigante abscedado benigno debido a su baja frecuencia clínica. Caso clínico: Paciente masculino de 53 años, con enfermedad actual desde agosto/2013 caracterizada por hiporexia, astenia, dolor en hipocondrio derecho, concomitantemente fiebre en 39°C y pérdida de peso de 12 Kg en 4 meses, siendo referido a este centro. Durante su hospitalización presenta cifras tensionales elevadas y palpitaciones. Se realiza ecosonograma abdominal en 2 oportunidades con hallazgos de lesión ocupante de espacio (LOE) en segmentos hepáticos V y VI y lesión parenquimatosa renal derecha grado II. Se realiza TAC abdomino-pélvica con doble contraste donde se evidencia LOE suprarrenal derecho de aspecto neoproliferativo; se solicitan catecolaminas en orina de 24 horas que reportaron elevadas. Se inicia α y β bloqueo con doxazosin y propranolol. Posteriormente se realiza intervención quirúrgica: adrenalectomía derecha con vaciamiento ganglionar y nefrectomía derecha. Macroscópicamente se observó tumor adrenal de 25 cm de diámetro, con contenido purulento fétido en su interior, cuyo cultivo reportó Salmonella sp. La biopsia concluyó en feocromocitoma quístico abscedado con ausencia de hallazgos sugestivos de malignidad. Conclusión: El feocromocitoma es un tumor neuroendocrino con una baja prevalencia, la mayoría son menores de 6 cm, existiendo pocos reportes de casos de feocromocitomas mayores de 20 cm y de lesiones abscedadas, ambas comúnmente asociadas a malignidad. El diagnóstico definitivo es histológico. El tratamiento es la resección quirúrgica.
Palabras claves: feocromocitoma gigante, feocromocitoma quístico, feocromocitoma abscedado, metanefrinas, adrenalectomía.
Giant pheochoromocytoma abscessed: A case report.
ABSTRACT
Objectives: To describe a case of a giant benign abscessed pheochromocytoma due to its low incidence. Clinical case: A fifty-three year old male patient, with current illness since august/2013, characterized by hyporexia, astenia, abdominal pain on right hypochondrium and 39°C fever with 12 kg weight loss in a 4 month period, was referred to this medical center. During hospital stay the patient presents elevated blood pressure and palpitations. An abdominal US is performed twice with the following findings: space-occupying lesion (SOL) on liver segments V and VI and a grade II right renal parenchymal lesion. A double contrast CT-Scan of the abdomen and pelvis is performed and reports a neo-proliferative right suprarenal mass. The 24-hour urinary catecholamine test result was high. α and β blocking with doxazosin and propranolol was initiated. Soon after he was operated: right adrenalectomy with lymph node resection and right nephrectomy. Grossly an adrenal tumor of 25 cm diameter is observed, with fetid, purulent inside content. Culture of purulent content reported Salmonella sp. Biopsy was concluded as cystic-abscessed pheochromocytoma with no suggestive findings of malignancy. Conclusion: Pheochromocytoma is a low prevalence neuroendocrine tumor. Most are less than 6cms, with few case reports of pheochromocytomas more than 20 cm and very few of abscessed lesions, both commonly associated with malignancy. The definitive diagnosis is histological. The treatment is surgical resection.
Key words: giant pheochromocytoma, cystic pheochromocytoma, abscessed pheochromocytoma, metanephrines, adrenalectomy.
Articulo recibido en: Septiembre 2014 Aceptado para publicación en: Enero 2015
INTRODUCCIÓN
El feocromocitoma es un tumor neuroendocrino productor de catecolaminas que se origina de las células cromafines del sistema simpático adrenal, con una incidencia de 3-8 casos por millón de habitantes, su prevalencia se estima en 1:1700 a 1:4500, sin diferencias en cuanto al sexo1. Puede ser de origen adrenal (75-80%) o extra-adrenal (20-25%), estos últimos también denominados paragangliomas. La mayoría de los feocromocitomas son benignos2.
Clínicamente sus síntomas se deben a la secreción de catecolaminas a la circulación sanguínea produciendo cefalea, palpitaciones, diaforesis y ansiedad, puede asociarse hipertensión arterial paroxística (48%) o persistente (29%)3. El diagnóstico se basa en la confirmación bioquímica del exceso hormonal para lo cual se pueden determinar los niveles de catecolaminas en plasma o en orina. Posterior a la confirmación de hiperfunción hormonal se debe localizar anatómicamente el tumor secretor de catecolaminas4.
El tratamiento es quirúrgico, con la resección del tumor previa estabilización hemodinámica con antagonistas α y bloqueantes β adrenérgicos4.
CASO CLÍNICO
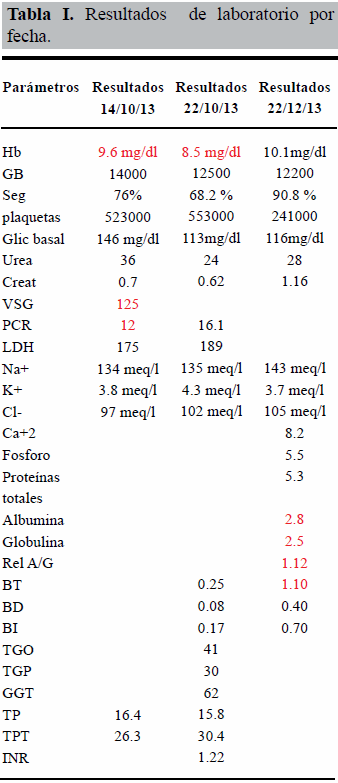
Paciente masculino de 53 años, con enfermedad actual (EA) desde agosto/2013 caracterizada por hiporexia, astenia, dolor abdominal opresivo en hipocondrio derecho irradiado a región lumbar ipsilateral, concomitantemente fiebre en 39°C y pérdida de peso de 12 Kg en 4 meses, siendo referido a este centro, ingresando a cargo de gastroenterología. Durante su hospitalización presenta cifras tensionales elevadas (150/90 mmHg) y palpitaciones al deambular. Se realizó ecosonograma abdominal en 2 oportunidades con hallazgos de: hepatomegalia, lesión ocupante de espacio (LOE) en segmentos hepáticos V y VI y lesión parenquimatosa renal derecha grado II. Se inició antibioticoterapia ante la sospecha clínica de absceso hepático (ampicilina/sulbactam) vía endovenosa por 14 días sin mejoría. Se realizó TAC abdomino-pélvica con doble contraste que reportó LOE suprarrenal derecho de aspecto neoproliferativo e hiperflujo en segmentos hepáticos VI y VII por compresión extrínseca (Figs. 1 y 2); fue trasladado al servicio de cirugía IV, se solicitan catecolaminas en orina de 24 horas que reportaron: catecolaminas totales: 1135 μg (VN: 25-312), epinefrina: 400 μg (VN: 0-20), norepinefrina 735 μg (VN: 0-90), dopamina 1224 μg (VN: 0-600), y ácido vanililmandelico: 28,4 mg (VN: 0-15). (Tabla 1).
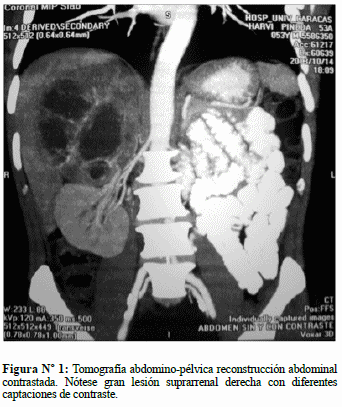

Fue evaluado por endocrinología, se inicia α y β bloqueo pre-operatorio con doxazosin a dosis de 2 mg vía oral cada 12 horas y propranolol 20 mg vía oral cada 8 horas respectivamente, aumentándose a las 72 horas las dosis a doxazosin 4 mg cada 12 horas y propranolol 40 mg cada 8 horas hasta alcanzar los síntomas clínicos de α y β bloqueo (normotensión y frecuencia cardiaca en valores normales) a las dos semanas de inicio del tratamiento. Antecedentes personales patológicos: niega patologías crónicas; antecedente quirúrgico: tendinoplastia del 3er dedo de mano derecha hace 20 años. Niega alergia a medicamentos. Antecedentes familiares: no contributorios. Hábitos psicobiológicos: tabáquicos: 5 paquetes/año; alcohólicos: cerveza los fines de semana (30 cervezas) desde los 15 años hasta la actualidad, cafeínicos e ilícitos niega. Al examen funcional refiere pérdida de peso de 12 kg en los últimos 4 meses, dolor abdominal y alza térmica como descritos en la EA, hábito evacuatorio diario (2 veces/día) y hábito miccional (5 veces/día) sin alteraciones. Al examen físico: TA: 150/90mmHg (acostado), 160/96 mmHg (sentado), FC: 84x (acostado), FC: 98x (sentado), FR: 20x, Temperatura: 38,3 °C, Peso: 58 Kg, Talla: 1,70 m IMC: 20 Kg/m2. Paciente en regulares condiciones generales, hidratado, eupneico. Piel: morena, ligeramente pálida. ORL: mucosa oral húmeda, edéntula parcial. Cuello: móvil, no doloroso, tiroides no visible ni palpable.
Cardiopulmonar: ruidos cardiacos rítmicos, normofonéticos, sin soplos ni 3er o 4to ruido, ruidos respiratorios presentes en ambos campos pulmonares, sin adventicios. Abdomen: ruidos hidroaéreos presentes, abdomen con leve asimetría dada por aumento de volumen de región subcostal derecha, blando, deprimible, con dolor moderado a la palpación en hipocondrio derecho y región lumbar ipsilateral, hepatometría 12/14/16 cms, sin evidencia de solución de continuidad en piel ni secreción purulenta. Extremidades: simétricas, eutróficas, sin edemas. Neurológico: consciente, orientado en 3 planos, sin signos de focalización neurológica.
En diciembre 2013 se realiza intervención quirúrgica: cirugía abierta, practicándosele adrenalectomía derecha con vaciamiento ganglionar, nefrectomía derecha, colecistectomía y biopsia incisional hepática. Macroscópicamente se observó tumor adrenal de 25 cm de diámetro, con contenido purulento fétido en su interior (Figs. 3 y 4). Durante acto quirúrgico presentó inestabilidad hemodinámica, requiriendo en el post-operatorio inmediato hemoderivados y nitroprusiato; permaneció 5 días en la unidad de terapia intensiva, con tendencia a la hipotensión por shock distributivo, posteriormente se traslada a sala general donde permaneció 3 semanas. Durante su estadía en sala estuvo con tendencia a la hipertensión arterial y frecuencia cardiaca normal a normal-alta, se mantuvo α y β bloqueo por dos semanas, posteriormente se inició descenso de dosis; egresó asintomático y hemodinámicamente estable. El cultivo de secreción purulenta reportó Salmonella sp. Biopsia reportó: feocromocitoma con signos de hemorragia, abscedación y necrosis, capsula tumoral sin infiltración, riñón y uréteres sin infiltración, ganglios linfáticos perihileares y de vena cava con hiperplasia sinusoidal y folicular reactiva libre de neoplasia. Biopsia incisional hepática con inflamación crónica granulomatosa necrotizante. No se realizó inmunohistoquimica.
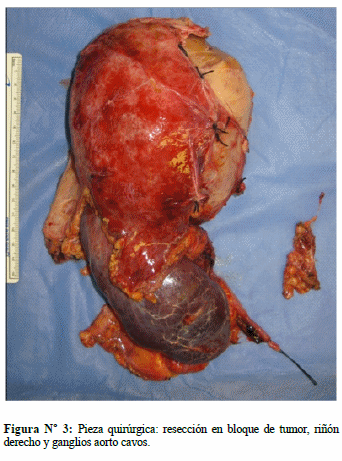
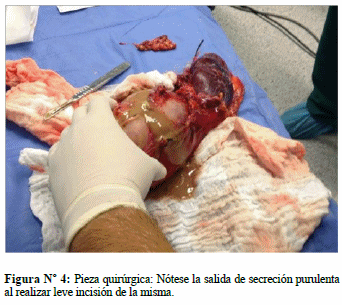
DISCUSIÓN
El feocromocitoma es un raro tumor vascular neuroendocrino que se origina de las células cromafines del sistema simpático adrenal. Su prevalencia se estima de 1:1700 a 1:4500, con una incidencia anual de 3-8 casos por millón de habitantes1.
Los feocromocitomas pueden ser de origen adrenal (80-85%) y extra-adrenal (15-20%), estos últimos también conocidos como paragangliomas. Aproximadamente el 30-35% de los feocromocitomas y paragangliomas son hereditarios formando parte de los síndromes de neoplasia endocrina múltiple tipo 2 (NEM 2), neurofibromatosis tipo 1 (NF-1), enfermedad de Von Hippel-Lindau tipo 2 (VHL 2) y paragangliomas familiares2.
La formación de abscesos puede ocurrir a una gran variedad de órganos, siendo estos más comunes en huésped inmunocomprometidos. Se ha reportado que la formación de abscesos en la glándula adrenal puede ser inducido por infecciones placentarias las cuales posteriormente pueden llevar a hemorragias adrenales, sin embrago los abscesos en el feocromocitoma son raros, existiendo solo 3 casos reportados en la literatura3 ; el primero de estos casos se reportó en 1954 en una paciente femenina de 29 años de edad con fiebre de 3 semanas de evolución e hipertensión arterial paroxística, aislándose el germen de Samonella Typhimurium4; el segundo caso fue reportado en el 2007, en un hombre de 34 años de edad quien cursó con clínica de fiebre, dolor lumbar, cefalea y palpitaciones paroxísticas con el diagnóstico de un feocromocitoma abscedado en la glándula adrenal derecha, cultivo de secreción de lesión glandular reportó Streptococcus Agalactiae5; el tercer caso fue reportado en el 2012 en una paciente femenina de 53 años diabética tipo 2 quien cursó con fiebre y fatiga progresiva de 1 semana de evolución, había presentado 2 semanas previas a su enfermedad actual episodio de hemorragia subaracnoidea secundaria a emergencia hipertensiva, bioquímicamente con leucocitosis, elevación de la proteína C reactiva (PCR), catecolaminas y metanefrinas plasmáticas y urinarias elevadas e imagen de TAC abdominal que mostraba una LOE suprarrenal derecho de 12×14 cms, se realizan 2 hemocultivos que reportan bacteriemia por Campylobacter fetus y posterior a la adrenalectomía del LOE se realiza cultivo de secreción purulenta fétida el cual evidencia el mismo germen (C. fetus), concluyéndose en una crisis del feocromocitoma secundaria a la formación de un absceso tumoral suprarrenal por bacteriemia3.
Hasta donde se tiene conocimiento y en la revisión realizada, el presente caso constituye el cuarto caso reportado en la literatura de feocromocitoma abscedado y el primer caso reportado cuyo germen aislado es Salmonella sp. Clínicamente sus síntomas se deben a la secreción de catecolaminas a la circulación sanguínea produciendo cefalea, palpitaciones, diaforesis, ansiedad, puede asociarse hipertensión arterial paroxística (48%) o persistente (29%)6. En el presente caso se evidenció un predominio de sintomatología infecciosa caracterizada por alza térmica probablemente en correlación con hallazgo de Salmonella sp, evidenciándose posteriormente en su hospitalización los síntomas y signos secundarios a hipersecreción hormonal adrenal. Esta presentación coincide con la referida por Sath y col (2011) quienes describen un caso clínico de un paciente masculino de 57 años de edad quien debuta con fiebre de origen desconocido, evidenciándose posteriormente lesión suprarrenal (feocromocitoma) de 4,5 cm de diámetro, no abscedada7; similar también al caso referido por Inoue y col (2006) quienes describen un paciente masculino de 34 años de edad que debuta con síntomas de fiebre y dolor lumbar, el cual no mejora con antibióticos, en quien posteriormente se realiza adrenalectomía laparoscópica evidenciándose lesión abscedada de 4 cm de diámetro con Streptococus agalactiae como germen aislado5, definiéndose así la heterogeneidad en la presentación clínica de esta patología.
El feocromocitoma puede ocurrir a cualquier edad, con igual distribución por género, aunque la edad de presentación puede predecir el fenotipo del tumor secretor de catecolaminas y la mutación genética subyacente, presentaciones a edades más tempranas se asocian a síndromes hereditarios y a mayor edad se asocia a presentación esporádica7; en el paciente presentado no se evidenciaron al interrogatorio antecedentes familiares sugestivos de presentaciones heredo-familiares, ni características al examen físico sugestivas de NEM-2, NF-1 ni VHL-2, coincidiendo esta presentación esporádica con las descritas para su grupo etáreo.
El diagnóstico se basa en la confirmación bioquímica del exceso hormonal, para lo cual se pueden determinar los niveles de catecolaminas en plasma o en orina, presentando la determinación de los productos de degradación de las catecolaminas (metanefrinas y normetanefrinas) mayor sensibilidad para detectar la hiperfunción hormonal, siendo el estándar de oro para el diagnóstico la determinación de los niveles plasmáticos libres de normetanefrina y metanefrina; si esta prueba es realizada por cromatografía líquida alcanza una sensibilidad del 96-100%, aunque presenta una baja especificidad (85-89%) por lo que está indicada en pacientes con alto pre-test para feocromocitoma; en aquellos pacientes con menor probabilidad de presentar la enfermedad se deberán solicitar inicialmente los niveles de metanefrina y normetanefrina en orina de 24 horas7. En este paciente se evidenció elevación significativa de todos los productos de degradación de las catecolaminas en muestra de orina de 24 horas corroborándose el diagnóstico bioquímico.
Una vez que el diagnóstico bioquímico se ha confirmado, se procede a la localización de la lesión. Aproximadamente 85% de estas lesiones se encuentran en las glándulas adrenales y el resto son de localización extra-adrenal (abdomenpelvis) 8. La tomografía axial computarizada abdomino-pélvica con contraste es el estudio de primera línea con una sensibilidad del 98%, seguida de la resonancia magnética por imágenes (RMI) con una sensibilidad del 89%; la tomografía permite diferenciar las lesiones tipo adenoma de los feocromocitomas, los hallazgos característicos de adenoma adrenal benigno en tomografía incluyen: un bajo valor de atenuación con menos de 10 unidades hounsfield (UH) con un rápido lavado del contraste (mayor al 50%) a los 10 minutos posteriores a la administración del mismo8. Los adenomas benignos son usualmente redondeados, con densidad homogénea, menores a 3 cm de diámetro y generalmente unilaterales, a diferencia de los feocromocitomas, los cuales suelen ser de densidad homogénea o heterogénea, sólidos, quísticos o necróticos, con una alta densidad al contraste (mayor de 10 UH) y un lavado del contraste menor al 50% a los minutos de la administración del mismo9. En los feocromocitomas extra-adrenales, metástasis y recidivas, la RMI presenta mayor sensibilidad que la TAC10. Si el tumor no puede ser localizado por su pequeño tamaño, localización u otra razón, se puede realizar una gammagrafía con metaiodobencilguanidina (131-I-MIBG), la cual tiene una sensibilidad del 81% y especificidad del 99% y en conjunto con los niveles de normetanefrina plasmática libre elevada alcanza una sensibilidad del 100%10. La tomografía de este paciente describe una lesión de gran tamaño (mayor de 20 cm en los cortes sagitales), no fue necesario la realización de estudios de imágenes más complejos; llamó la atención el volumen de la lesión y la captación heterogénea del contraste, hallazgo compatible con lo encontrado en post-operatorio de feocromocitoma gigante abscedado.
Los feocromocitomas gigantes son tumores poco frecuentes, con pocos casos descritos en la literatura y comúnmente asociados a malignidad, como el reportado por Soufi y col (2009) quien describe a un paciente masculino de 17 años de edad con tumor suprarrenal con aspecto quístico de 21 cm de diámetro, macroscópicamente la pieza postquirúrgica presenta necrosis central y adherencia al riñón derecho, la biopsia e inmunohistoquímica son sugestivas de feocromocitoma maligno, el paciente fallece a los dos años de la cirugía; estos autores concluyen que el tamaño del tumor, excesiva secreción hormonal e infiltración local son predictores de malignidad, hallazgos que contrastan con lo descrito en el presente caso11.
La definición de malignidad para el feocromocitoma ha sido difícil de comprender, ya que esta no se basa hasta el momento de la presente revisión, en criterios histológicos como ocurre con la mayoría de los tumores, según la OMS la definición rigurosa de malignidad requiere la presencia de metástasis en áreas donde no es posible encontrar tejido cromafin, siendo las zonas de metástasis más frecuentemente descritas: esqueleto axial, ganglios linfáticos, hígado, pulmón y riñón; quedando bajo este concepto difícil establecer casos de recidiva local o de un tumor primario asincrónico los cuales cursan con lesiones metastásicas, elevación de catecolaminas y síntomas clínicos similares12,13.
En el 2002 Thompson realiza un estudio retrospectivo donde evalúa las características clínicas, histopatológicos e inmunohistoquimica de 100 piezas patológicas del instituto de patología de las fuerzas armadas de Washington DC, (EEUU) correspondientes a feocromocitomas de aquellos pacientes quienes completaron el seguimiento post- quirúrgico hasta 10 años, el objetivo fue evidenciar si existía alguna correlación entre parámetros clínicos, bioquímicos, histológicos e inmunohistoquimicos con malignidad; posterior a la realización del estudio determinan que no existen factores clínicos, comportamiento bioquímico ni hallazgos inmunohistoquímicos que predigan malignidad, evidencian una mayor asociación de malignidad con la presencia de determinados parámetros histológicos y proponen un sistema de puntuación o score denominado PASS por sus siglas en inglés (Pheochromocytoma of the Adrenal Gland Scaled Score) para el cual tomaron en cuenta los siguientes parámetros histológicos: 1.-lesiones grandes o con crecimiento difuso (>10% del volumen), 2.- necrosis tumoral confluyente o central, 3.- alta celularidad, 4.- presencia de un mismo tipo celular en el tejido, 5.- ejes de células tumorales (aun si es focal), 6.- presencia de más de 3 mitosis por campo en microscopía de alta resolución, 7.- mitosis atípicas, 8.- extensión dentro del tejido adiposo, 9.- invasión vascular, 10.- invasión capsular, 11.- pleomorfismo, 12.- hipercromasia, teniendo los hallazgos 1 al 8 un valor de 2 puntos cada uno y los restantes un valor de 1 punto cada uno, encontrando un comportamiento biológicamente agresivo en aquellos tumores con un score ≥ 4 puntos14. Este score deberá ser validado en estudios clínicos con mayor población.
El diagnóstico de malignidad en el feocromocitoma es realizado prospectivamente en el seguimiento del paciente, siendo malignos aquellos casos en los cuales aparecen lesiones metastásicas en las áreas descritas, no existiendo estadísticas reportadas de la incidencia del mismo. Por la ausencia de lesiones metastásicas en los ganglios linfáticos evaluados y la ausencia de metástasis hepática en la biopsia realizada en nuestro paciente se puede catalogar como benigno para el momento de esta revisión, quedando pendiente el seguimiento de este caso para descartar aparición de lesiones metastásicas las cuales pueden aparecer de 5 a 10 años después de la cirugía1.
En el 2011 De Wailly y col realizaron un estudio retrospectivo tomando como muestra aquellos pacientes operados de feocromocitoma entre 1993-2009, clasificando a los pacientes en 3 grupos: grupo 1: pacientes con metástasis documentadas, grupo 2: pacientes con feocromocitoma benigno con PASS > 4 y grupo 3: pacientes con feocromocitoma benigno con PASS < 4, evaluaron la asociación entre puntaje de PASS, tamaño tumoral, índice mitótico (Ki-67) y presencia de la proteína S-100; encontraron que el tamaño y peso tumoral esta correlacionado con malignidad (p<0,05) y que aquellos pacientes con un índice mitótico > 4% y ausencia de proteína S-100 ameritan un seguimiento más estrecho por riesgo de aparición de metástasis15. En el presente caso no se realizaron pruebas inmunohistoquimicas y el seguimiento actual no ha evidenciado alteraciones sugestivas de malignidad debiéndose esperar hasta 10 años para descartar la posibilidad de metástasis.
Una vez localizada la lesión se planifica la cirugía, que es el tratamiento de elección5, existiendo una tendencia actual a realizar abordaje laparoscópico12, dependiendo del tamaño y características de la lesión (lesiones menores de 6 cm); en este paciente se decidió realizar cirugía abierta considerando el tamaño de la lesión.
En conclusión, el feocromocitoma es un tumor neuroendocrino con una prevalencia de 3-8 casos por millón de habitantes, la mayoría son menores de 6 cm, existiendo pocos reportes de casos de feocromocitomas mayores de 20 cm y de lesiones abscedadas, ambas comúnmente asociadas a malignidad. Existe una heterogeneidad en la presentación clínica de estas lesiones las cuales pueden debutar con síntomas generales e inespecíficos, síntomas infecciosos (síndrome febril subagudo o fiebre de origen desconocido) o con síntomas de compresión abdominal por efecto de masa, además de los síntomas clásicamente descritos por hiperestimulación del sistema α y β adrenérgico. Los síntomas infecciosos deben hacer sospechar la posibilidad de un germen sobreinfectando la lesión y en el cual el tratamiento antibiótico no produce mejoría, en probable correlación con la dificultad para la llegada del antibiótico a dicha zona. Independientemente de las características macroscópicas de la lesión e infiltración de estructuras contiguas, el diagnóstico definitivo es histológico. El tratamiento es la resección quirúrgica, con buen pronóstico a corto y largo plazo en el caso de feocromocitomas benignos.
REFERENCIAS BIBLIOGRÁFICAS
1. Tsirlin A, Sharma R, Kansara A, Gliwa A, Banerji M. Pheocrocytoma: A review. Maturitas 2014;77:229– 238. [ Links ]
2. Karasek D, Shah U, Frysak Z, Stratakis C, Pacak K. An update on the genetics of pheochromocytoma. J Hum Hypertens 2013;27:141-147. [ Links ]
3. Abe I, Nomura M, Watanabe M, Shimada S, Kohno M, Matsuda Y, Adachi M, Kawate H, Ohnaka K, Takayanagi R. Pheochromocytoma crisis caused by Campylobacter fetus. Int J Urol 2012;19:465-467. [ Links ]
4. Giel CP. An abscess formation in a pheochromocytoma; report of a case due to Salmonella typhimurium. N Eng J Med 1954;251:980-982. [ Links ]
5. Inoue R, Hisasue S, Kunishima Y, Masumori N, Itoh N, Tsukamoto T. Pheochromocytoma with abscess. Int J Urol 2007;14:644-646. [ Links ]
6. Bravo E, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev 2003;24:539-553. [ Links ]
7. Sath M, Karra R, Mark D. Pheocromocytoma presenting as fever of unknow origin. Am J Med 2012;124:5-6. [ Links ]
8. Eisenhofer G, Timmers HJ, Lenders JW. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab 2011; 96:375-384. [ Links ]
9. Chen H, Sippel RS, ODorisio MS, Vinik AI, Lloyd RV, Pacak K, North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 2010;39:775-783. [ Links ]
10. Ctvrtlika F, Korandab P, Tichyc T. Adrenal disease: a clinical update and overview of imaging. A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2014;158:23-34. [ Links ]
11. Soufi M, Mohammed K, Lahlou S, Benam R, Jalil M, Abdelhamid E, Mohammadine E, Ahmed T, Abdelatif S, Bouziane C. Giant malignant cystic pheochromocytoma: a case report. Indian J Surg 2012;74:504-506. [ Links ]
12. - Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Williams textbook of Endocrinology. 12th ed. Philadelphia. Elsevier; 2011:515-530.
13. DeLellis RA, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumours of endocrine organs. Lyon, France: IARC Press; 2004. World Health Organization Classification of Tumours; vol 8.
14. Thompson LD. Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002; l26:551-566. [ Links ]
15. De Wailly P, Oragano L, Radé F, Beaulieu A, Arnault V, Levillain P, Kraimps JL. Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg 2012;397:239–246. [ Links ]
