











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
Permalink
INTRODUCCIÓN
La linfohistiocitosis hemofagocítica (HLH), descrita por primera vez en 1939 como reticulosis medular histiocítica por Scott y Robb-Smith 1. La HLH es un trastorno raro que se distingue por hiperinflamacion sistémica descontrolada y grave, debido a la activación anómala y persistente del sistema inmune (linfocitos T citotóxicos y las células asesinas naturales) debido a causas primarias o secundaria 2,3.
La infección por el virus de Epstein Barr (VEB), es la causa infecciosa más frecuente relaciona a la HLH 3. Esta infección de los linfocitos T citotóxicos CD8+ por el VEB, induce la activación incontrolada y su actividad defectuosa con aumento de la secreción de citocinas inflamatorias y activación de los macrófagos mediado por el interferón-gamma (IFN-γ) provocando un círculo vicioso de inflamación sistémica 3,4. Desde el año 1989 hasta la publicación del ensayo terapéutico HLH-2004, se han reportado más de 700 pacientes en todo el mundo, con una incidencia de 1 a 225 por cada 300.000 nacidos vivos que varía de región geográfica a otra 3,5.
Este síndrome tiene características clínicas y analíticas como: fiebre, esplenomegalia, erupciones diseminadas en la piel, alteraciones en el sistema nervioso central (SNC), edema, ictericia, citopenias, coagulopatía, lactato deshidrogenasa (LDH) elevada, hipertransaminasemina, elevación de biomarcadores de la inflamación (ferritina, triglicéridos), con progresión a fallo multiorgánico que en la mayoría de los casos provocan la muerte 3,6. Para el diagnostico en primer lugar se debe tener alta sospecha clínica debido a que no único examen diagnóstico para la HLH, sin embargo, la Sociedad de Histiocitosis planteó 8 criterios en el ensayo internacional HLH-2004 siendo modificado en el año 2007.
Estos criterios se dividen en dos clínicos (fiebre y esplenomegalia), y 6 analíticos (función baja o ausente de las células asesinas naturales (NK), citopenias, hipertrigliceridemia, hipofibrinogenemia, ferritina elevada, hemofagocitosis en el mielograma y CD25 soluble elevado (receptor alfa de interleucina 2 (IL2Rα)) 4,7. El tratamiento va dirigido a interrumpir y controlar la hiperinflamación, la tormenta de citocinas y destruir los macrófagos activados.
En la actualidad se utiliza el protocolo HLH 2004 que consiste en administrar una combinacion de quimioterapia proapoptótica e inmunosupresores contra células T activadas y hacia los histiocitos, logrando una remisión completa en los 90% de los casos 4,8. Este reporte de caso tiene como objetivo instruir a los médicos de primera línea sobre las características clínicas y de laboratorios de la HLH y terapéutica oportuna.
MATERIALES Y MÉTODOS
Revisión de historial clínico de la paciente y de literatura médica científica. El consentimiento informado por escrito se obtuvo en el departamento de docencia del Hospital de niños Dr. Roberto Gilbert Elizalde antes de su publicación.
Descripción del caso
Se presenta el caso de una lactante mayor femenina, sin antecedentes personales ni familiares de importancia, fue atendida en el Hospital IESS Ceibos y posteriormente en la unidad de cuidados intensivos del Hospital de Niños Dr. Roberto Gilbert Elizalde en Guayaquil durante los meses de abril y mayo de 2024.
La paciente presentó un cuadro clínico de 10 días de evolución caracterizado por fiebre de hasta 40°C, deposiciones diarreicas de 2 a 3 por día sin moco ni sangre, y progresión a somnolencia y dificultad respiratoria severa en las últimas horas.
A su llegada a la unidad de cuidados intensivos pediátricos, se encontraba en estado de choque, lo que requirió intubación rápida y soporte con aminas vasopresoras. El examen físico inicial mostró a una paciente pálida, visiblemente deshidratada, con un tiempo de relleno capilar prolongado de 4 segundos y taquicardia significativa. Presentaba un rash petequial confluente eritematoso en cara, tórax y abdomen, gran distensión abdominal con hepatosplenomegalia, y el hígado extendido hasta el flanco derecho.
Los estudios de laboratorio iniciales (Tabla 1) del 26 de abril de 2024 mostraron resultados alarmantes, incluyendo anemia normocítica normocrómica arregenerativa, leucopenia severa, neutropenia severa, linfopenia moderada, trombocitopenia severa, hipertrigliceridemia, hiperferritinemia e hipofibrinogenemia. Los marcadores inflamatorios estaban extremadamente elevados, con una PCR de 23.9 mg/dL, LDH de 3322 U/L, y ferritina de 3028 ng/mL. Los niveles de triglicéridos eran de 698 mg/dL, sugiriendo un estado inflamatorio sistémico severo. Las enzimas hepáticas (TGO 1337 U/L, TGP 545 U/L) y bilirrubinas totales (2.88 mg/dL) confirmaron el compromiso hepático significativo. Además, el tiempo de protrombina prolongado y la no coagulación en TTP indicaban coagulopatía. Estudio de tomografía simple de abdomen con evidencia de hepatomegalia y ascitis. Las pruebas virológicas revelaron una alta carga viral de Epstein-Barr (7479014 copias/mL en sangre y 320910 en médula ósea), junto con una infección concomitante por CMV (250 copias), mientras que las pruebas para HIV y tuberculosis fueron negativas. En el mielograma se evidenció hipocelularidad medular con hemofagocitosis evidente, consolidando el diagnóstico de linfohistiocitosis hemofagocítica (HLH) secundaria a infecciones por VEB y CMV (Figura 1 y 2).
Tabla 1 Resultados de exámenes iniciales
| HEMOGRAMA | VALOR |
|---|---|
|
|
1140/mm³ |
|
|
15.8% (180/mm³) |
|
|
78.9% (900/mm³) |
|
|
4.4% (50/mm³) |
|
|
0% |
|
|
5.8 g/dL |
|
|
16.6% |
|
|
78.3 fl |
|
|
27.4 pg |
|
|
35000/mm³ |
| MARCADORES INFLAMATORIOS | VALOR |
|
|
23.9 mg/dL |
|
|
3322 U/L |
|
|
50 mg/dL |
|
|
3028 ng/mL |
|
|
698 mg/dL |
| FUNCIÓN HEPÁTICA | VALOR |
|
|
1337 U/L |
|
|
545 U/L |
|
|
278 U/L |
|
|
2.88 mg/dL |
|
|
2.28 mg/dL |
|
|
0.60 mg/dL |
| FUNCIÓN RENAL | VALOR |
|
|
28.2 mg/dL |
|
|
0.17 mg/dL |
|
|
2.8 mg/dL |
| COAGULACIÓN | VALOR |
|
|
15.2 seg |
|
|
1.4 |
|
|
No coagula |
| INMUNOLOGÍA Y VIROLOGÍA | |
|
| |
| MIELOGRAMA | |
|
| |
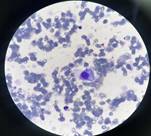
Figura1. Mielograma demostró hipocelularidad medular con hemofagocitosis evidente, sin la presencia de células inmaduras.
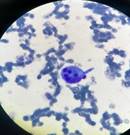
Figura 2 Mielograma demostró hipocelularidad medular con hemofagocitosis evidente, sin la presencia de células inmaduras.
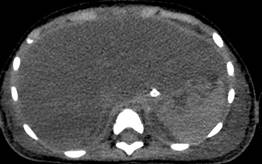
El manejo inicial incluyó soporte intensivo con ventilación mecánica, vasopresores, transfusiones y tratamiento antibiótico empírico. La sospecha de intoxicación por paracetamol, debido a la administración excesiva referida por los familiares, condujo al inicio de tratamiento con N-acetilcisteína.
A pesar de las intervenciones intensivas, la condición de la paciente continuó deteriorándose. Se inició el tratamiento urgente para linfohistiocitosis hemofagocítica (HLH) basado en el protocolo HLH-2004 de la Histiocyte Society, que incluyó dexametasona, inmunoglobulina y 50% de etopósido, además de profilaxis. No obstante, la paciente evolucionó a fallo multiorgánico irreversible y falleció a los doce días de estancia en la unidad de cuidados intensivos, a pesar de las intervenciones y el soporte vital avanzado brindado.
Tabla 2 H score para hemofagocitosis en la paciente
| H score para hemofagocitosis en la paciente | ||
|---|---|---|
| HALLAZGO | CLASIFICACIÓN | PUNTAJE |
| Fiebre |
|
|
| Organomegalia |
|
|
| Citopenia |
|
|
| Triglicéridos |
|
|
| Fibrinógeno |
|
|
| TGO |
|
|
| Ferritina |
|
|
| Hemofagocitosis |
|
|
| Inmunosupresión |
|
|
| TOTAL | 304 | |
| Un H-Score > 250 corresponde a una alta probabilidad de HLH y síndrome de activación de macrófagos. | ||
DISCUSIÓN
El término hemofagocitosis se define como la absorción, por los macrófagos activados, de leucocitos, glóbulos rojos y plaquetas en diferentes tejidos del organismo, produciendo una tormenta de citocinas e inflamación desenfrenada 9. La linfohistiocitosis hemofagocítica es un subconjunto de la familia más amplia de histiocitosis y neoplasias de los linajes de macrófagos y células dendríticas, es una entidad rara y de mortalidad considerable, alrededor del 50 - 75%.
Se presenta más comúnmente niños menores de 1 año en un 70 - 80 % de los casos con una tasa de incidencia de 1 caso por cada millón de recién nacidos al año con variaciones según la región geográfica (8). Se caracteriza por inflamación sistémica descontrolada mediado por citocinas inflamatorias, debido a la activación anómala del sistema inmunitario 3,10.
Se presenta fiebre, espenomegalia, elevación de biomarcadores inflamatorios y alteración en la coagulación por lo que alcanzar el diagnóstico definitivo es un verdadero desafío debido a la superposion clínica y analítica con otras enfermedades como las neoplasias malignas, trastornos autoinmunes y la sepsis 10. Es de gran importancia establecer un diagnóstico diferencial con estas condiciones debido a que el manejo terapéutico difiere. El paciente expuesto era una lactante que debuto con fiebre, exantema generalizado esplenomegalia, citopenias y reactantes de fase aguda elevados. Se realizo mielograma donde se evidenció la presencia de hemofagocitos y ausencia de linfoblastos. El protocolo de la Sociedad del Histiocito plantea 8 criterios poniendo énfasis en cumplir 5 criterios para confirmar el diagnostico 2. En el paciente presentado se establecieron 6 criterios para linfohistiocitosis hemofagocitica (fiebre, esplenomegalia, ferritina elevada, niveles bajos de fibrinógeno y en mielograma con presencia de hemofagocitos).
En nuestro hosptal no disponemos de reactivos para medir la actividad del receptor de IL-2 soluble y de las células asesinas naturales (NK). El virus de Epstein Barr (VEB), un herpes virus que produce infecciones leves en la mayoria de los casos, pero en raras ocasiones puede infectar células T y células asesinas naturales (NK), impulsar la linfoproliferación de células T/NK y asociarse a HLH. Los pacientes HLH y altas cargas de ADN del VEB en sangre periférica (ADN del VEB) deben clasificarse de la siguiente manera: HLH familiar asociada al VEB, HLH no familiar asociada al VEB no neoplásico, HLH asociada a neoplasia como manifestación clínica del VEB sistémico T/NK activo crónico (CAEBV), y HLH asociada a neoplasia como manifestación clínica de un linfoma de células T / NK del VEB 2.
La carga de ADN del VEB se correlaciona con los niveles de los tiempos de protrombia (TP) y tromboplastina parcial (TTP), debido expresión del factor tisular con gran consumo de los factores de la coagulación y provocando aumento de la actividad fibrinolítica y caída de la síntesis de factores de coagulación 11.
Una vez realizado el diagnostico de HLH se realizó el abordaje la etiología con determinación de carga viral de VEB con reporte de carga viral superior a 7 millones de copias en sangre periférica con coagulopatía de consumo debido a que el TTP no coagulaba. Se catalogó como HLH no familiar asociada al VEB no neoplásico.
El objetivo del tratamiento de la HLH controlar la hiperinflamación y revertir la respuesta inmunológica deletérea y desregulada, utilizando el protocolo terapéutico HLH 94 que se basa en agentes inmunosupresores (dexametasona), y mielosupresores (inhibidor de la epipodofilotoxina topoisomerasa-II etopósido) (8). En el protocolo HLH 2004 se recomienda el uso de ciclosporina, sin embargo, aún no se han publicado los resultados definitivos de este estudio. La eficacia de la terapéutica se evalua como respuesta completa, parcial, sin respuesta. En nuestro paciente se utilizó dexametasona y etopósido sin repuesta favorable evolucionando con falla multiorgánica y desenlace fatal.

