









 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCTION
Every hour, billions of cells die in us, and our tissues do not shrink because of a natural regulation whereby cell death is balanced by cell division. It is necessary to control both death and cell division in differentiated cells to balance the different cell populations, avoiding affecting the adjacent cells 1.
The process in which cells eliminate themselves in a controlled manner is called programmed cell death. Programmed cell death plays an important role during embryonic development, maintaining tissue homeostasis in the body and eliminating damaged cells 2. In contrast, excessive or defective cell death contributes to a broad spectrum of human pathologies. Low-rate cell death can result in the formation of cancer and autoimmune diseases 3, whereas high-rate cell death can result in neurodegenerative disease, immunodeficiency, or muscle atrophy 4.
Knowledge of specific/differential proteomic expression in each cell death is essential for the early detection, diagnosis, and prognosis of cell-death-related diseases. This knowledge is also crucial for the use of more precise and personalized pharmacological treatments 5. Cell death can be divided into three groups: programmed, regulated, and accidental 6. Programmed cell death is present in embryonic development and tissue homeostasis, such as apoptosis and necroptosis. Regulated cell death is that which, programmed or not, can be induced or inhibited by a specific molecular mechanism through pharmacology or genetic interventions, for example, the release of neutrophil extracellular traps (NETs), a regulated form of neutrophil cell death known as NETosis, modulates neutrophil toxic effects. Accidental cell death is triggered by external physical conditions, such as ischemia, freeze-thaw cycles, or high concentrations of pro-oxidants, an example of this type of death are oncosis and necrosis 6. Two mechanisms of programmed cell death are distinguished: apoptotic cell death, dependent on caspases such as extrinsic and intrinsic apoptosis, and non-apoptotic cell death, independent of caspases, such as autophagy and necroptosis 7.
MATERIALS AND METHODS
Search strategy
The present study is a narrative literature review comprising scientific studies conducted between May and July 2023 that sought to group and describe caspase-dependent and caspase-independent programmed cell death, describing the molecular mechanisms of apoptosis, necroptosis, and autophagy. The bibliographic search was carried out in the following electronic databases: Medline (PubMed), SciELO, Scopus, Science Direct, Cinahl, EMBASE-Excerpta Medica Data Base, LILACS, Google Scholar, Dialnet, and Cochrane Library Plus. The key-words used for the search were: programmed cell death, apoptosis, caspases, caspase inhibitory proteins, mitochondrial / intrinsic pathway, extrinsic pathway, necroptosis, autophagy, phosphatidylserine, FAS (APO-1/CD95), tumor necrosis factor (TNF) receptor type 1 (TNF-R1) and TNF-related apoptosis-inducing ligand (TRAIL) and cytochrome C, linked by the Boolean operators “AND” and “OR”. Additional records were gleaned by conducting a ‘snowball’ search, checking the reference lists of publications eligible for full-text review, and using ResearchGate to identify potential articles not included in the databases used in the study.
Inclusion and exclusion criteria
The following inclusion criteria were applied to select the articles: (1) Access to the full text; (2) be a review, clinical trial, observational study, or case report/study; (3) identify caspase-dependent and caspase-independent programmed cell death; (4) describe the molecular mechanisms of apoptosis, necroptosis, and autophagy; (6) studies whose publication date is from the beginning of the databases until July 2023; (6) languages were restricted to English, German, French, Italian, Spanish, and Portuguese. The exclusion criteria applied were: (2) Publications not related to programmed cell death and/or describe its molecular mechanisms; (2) duplicate documents.
Data extraction
After searching the databases for studies, the search titles were checked to identify duplicates and possible publications to add. After reading the abstract, a full-text review of the selected studies was performed. Two reviewers (D.F.-L. and J.S.-C.) scrutinized and synthesized data from all selected studies into a comprehensive table using a standardized data extraction. A third reviewer (B.S.) resolved all disagreements between them.
RESULTS AND DISCUSSION
Programmed Cell death
Apoptotic cell death functions individually and selectively and is executed through a highly stereotyped series of biochemical events that ensure rapid non-inflammatory cell elimination. For this reason, the apoptotic physiological process is produced and characterized by decreased cell size, vesicle formation, and condensation of the nucleus. This series of transformations regulates the control of morphogenesis and organogenesis during embryonic development, in addition to tissue homeostasis in adult organisms 8.
Non-apoptotic cell death is usually described as a secondary mechanism for deficient apoptotic conditions. However, it is also possible that non-apoptotic programmed cell death mechanisms may function in first-order lines; for example, autophagic cell death is carried out during metamorphosis in insects. This autophagic cell death eliminates a tissue in its entirety 9.
In 1972, Kerr et al. 8 coined the term apoptosis to differentiate it from the death of natural origin, necrosis. The word comes from Greek, which refers to the leaves that fall from the trees or the petals that fall from the flowers. The prefix apo means “distance, outside or part” and ptosis “fall,” which literally means “fall from”. Apoptosis is associated with caspases, a family of cysteine proteases that control not only apoptosis but also proliferation, differentiation, cell shape, and cell migration.
Apoptosis is a form of programmed cell death 10, which occurs because of tissue and cell aging or in response to different external agents such as ionizing radiation and chemotherapeutic agents 10. It can be considered a process that facilitates the elimination of defective cells, so the alteration in the regulation of genes involved in cell death by apoptosis can cause and be associated with the development of different neoplasms, autoimmune diseases, viral infections, and neurodegenerative diseases 11. Apoptosis is an active process where the cells react and execute their death, programmed, by themselves 10. Apoptotic death is considered when a cell has lost its individuality or reached a “point of no return” at which the cell definitively loses its function. Apoptotic cell death triggers two stages. In the first stage, biochemical mediators attempt to repair a damaged cell. If they fail, the cell enters the second stage or execution phase, where structural changes occur that lead the cell to death12. These structural changes affect the cell membrane, intracytosolic organelles, and the nucleus 8. The cytoskeleton collapses, the nuclear envelope disassembles, redistributing the nuclear pores, the nuclear protein is altered, and the nuclear chromatin condenses and fragments, becoming dense clumps, with an electrophoretic “ladder” pattern, which migrate towards the nuclear membrane that shapes. DNA and RNA cleavage, due to the activation of Ca2+ and Mg2+ dependent endonucleases that cleave genomic DNA through the internucleosomal spaces 13. Also in the mitochondria, DNA degrades, and the endoplasmic reticulum loses its envelope; its cisterns widen and fuse 10. The phospholipids of the cell membrane change orientation and the phosphatidylserine residues are exposed to the external environment; the fragmentation of the phospholipids is induced because of these disruptions, the integrity of the plasma membrane is lost, and the mitochondrial membrane potential decreases. On the surface of the plasma membrane, fragments of membrane-enclosed cytoplasm called apoptotic bodies protrude and are shed, which are cytoplasmic remnants surrounded by a membrane such that they are rapidly engulfed by an adjacent cell or macrophage when released into the external environment, without causing an inflammatory response 12,14,15.
Caspases
Cysteine aspartate-specific proteases (Caspases, EC 3.4.22.-) are synthesized as inactive 30-50 kDa precursors (zymogens) that have three domains: an amino-terminal domain (prodomain), a domain that will result in a large subunit (p20) containing the active site, and another domain that will end in a smaller subunit catalyst (p10) C-terminus 16 (Fig. 1). In the presence of appropriate stimuli, a proteolysis process occurs between the domains, generating the active fragments. There are two types of caspases: initiator caspases (Caspase-2, 8, 9, and 10) that are activated in response to signs of stress or cell damage and that protect and activate effector caspases (Caspase-3, 6, and 7), these will oversee the direct proteolysis of different substrates that will lead to the death of the cell. One of their first identified substrates was Poly ADP-ribose polymerase (PARP) 17,18.
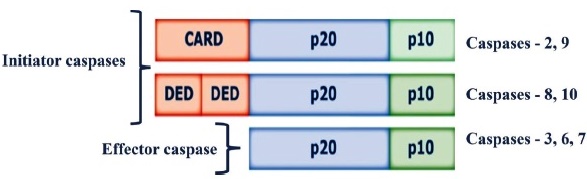
CARD: caspase recruitment and activation domain; DED: Death Effector Domain; p20: large subunit (p20); p10: small subunit (p10).
Caspases contain three domains: an N-terminal prodomain, a large subunit (p20) containing the active center with cysteine within a conserved QACXG motif, and a small subunit (p10) at the C-terminus.
Fig. 1 Structure of Caspases.
Initiator caspases present in their N-terminal region one or two essential adapter domains for their function. In contrast, effector caspases do not have these domains. There are two fundamental ways caspases can be activated (intrinsic and extrinsic pathways of caspase-dependent apoptosis), but although both pathways converge on effector caspases, they require different caspases to initiate the process. Thus, the activation of the extrinsic pathway mainly causes the recruitment of Caspase-8, and the intrinsic pathway principally causes the recruitment of Caspase-9 18,19.
During the process of apoptosis, there is a massive activation of caspases, which specifically cut proteins in cysteine residues located near aspartic acid. Caspases initiate a cascade of events that converge into a common effector caspase pathway that leads to the execution of apoptosis 12,20. The apoptotic machinery of the cytoskeleton has inactive precursors or initiating procaspases 8-10 that are activated by proteolytic cleavage and are catalyzed by other already active caspases; here, the process remains reversible. When activated, the initiator procaspases and cell-specific target proteins cleave and activate the executor procaspases (3, 6, and 7). From Caspase-3, the process is irreversible 17,19.
Apoptosis constitutes a complex series of positive and negative events with multiple positive and negative regulators and is integrated into other critical intracellular pathways such as cell cycle progression, phosphorylation-mediated signals, and DNA damage repair 6. Apoptosis is carried out mainly by two alternative pathways of apoptosis induction divided into i) apoptosis mediated by death receptors expressed on the cell surface or extrinsic pathway; ii) apoptosis mediated by the mitochondrial or intrinsic pathway 18. Signaling by both pathways induces the activation of caspases, and each pathway uses its own initiator caspases and activation complex 12,20.
Inhibitors of Apoptosis Proteins (IAPs)
Inhibitors of apoptosis proteins (IAPs) could inhibit apoptosis by selectively binding and inhibiting Caspase-3 and Caspase-7, but not Caspase-8. IAPs block the caspase cascade and inhibit cell death in response to proapoptotic stimuli 21. There are currently eight protein members of this family, but two of them, survivin and X-linked inhibitor of protein apoptosis (XIAP), are particularly interesting. In this sense, survivin is the only IAP associated with the mitotic spindle22. Its expression depends on the cell cycle 23. It has a double function since it inhibits apoptosis through direct and indirect interaction with caspases and regulates the cell cycle 24. Survivin is expressed in embryonic tissue and is overexpressed in tumor cells, associated with resistance to chemotherapy, but not in normal adult tissues 24,25. XIAP is possibly the best-studied IAP both at the structural level and at the level of its mechanism of action 26.
Furthermore, XIAP is the only member of this family that can inhibit both effector and initiator caspases. XIAP is frequently elevated in tumor cells, leading to resistance to chemotherapy 25. Therefore, XIAP is an optimal therapeutic target based on its functions; moreover, the inhibition of XIAP restores cellular chemosensitivity 27,28.
Caspase-dependent pathways of apoptosis
Extrinsic pathway: recipients of death
The extrinsic pathway is activated by ligands of the family of Tumor necrosis factor (TNF) proteins. Some ligands can induce apoptosis; when they bind to their receptors, they trigger caspase activation and initiate apoptosis 29. Death receptors are characterized by having cysteine-rich extracellular domains. They all have in common a death domain (DD) domain in the cytoplasmic region. In general, the binding of ligands to death receptors induces their trimerization, and subsequently, the DD domains recruit adapter molecules that will activate Caspase-8 and, when activated, activate Caspase-3 30. Extrinsic apoptosis is related to death receptors on the plasma membrane, such as phosphatidylserine (PS), FAS (APO-1/CD95), TNF receptor type 1 (TNF-R1), and TNF-related apoptosis-inducing ligand (TRAIL) 31.
Phosphatidylserine (PS)
In cells, the negatively charged PS is only localized to the cytosolic side of the lipid bilayer of the plasma membrane, but when it is translocated to the outer monolayer of the cell, it acts as an “eat me” signal, so it is considered as a marker of extrinsic apoptosis. In addition to expressing signals on the cell surface that stimulate apoptosis, PS also blocks inflammation in the phagocytic cell by inhibiting the production of cytokines proinflammatory signaling proteins 32. It must be considered that apoptotic cells must not only activate the signals that induce cell death but also inactivate or lose the death signals 12.
Fas (APO-1/CD95)
Fas (APO-1 / CD95) is ubiquitously expressed on the cell’s surface as a membrane protein of 40 kDa, which is highly expressed in T lymphocytes and activated natural killer (NK) cells 33. The activation of Fas at the cell surface is done by binding the Fas ligand (FasL) to the surface of a cytotoxic lymphocyte. The death domains of the cytosolic tails of Fas death receptors recruit adapter proteins, which in turn recruit procaspase initiators such as procaspase-8, procaspase-10, or both, forming the death-inducing signaling complex (DISC) 34. In practice, once activated in the DISC, the initiator caspases activate the next executor procaspases in the cascade, inducing apoptosis. This pathway begins with the formation of the DISC in which an adapter molecule called Fas associated death domain (FADD) and procaspase-8 intervene. FADD binds to Fas through their respective DD domains and to procaspase-8 through a death effector domain (DED). The oligomerization of procaspase-8 in the DISC complex results in the activation of Caspase-8 and subsequent activation of other caspases. Depending on the cell type, Caspase-8 can directly activate Caspase-3 or proteolyze the carboxy-terminus of BH3 interacting domain death agonist (Bid), a proapoptotic protein from the Bcl-2 family. Translocation of the truncated form of Bid into the mitochondria will activate the mitochondrial pathway 35,36 (Fig. 2).
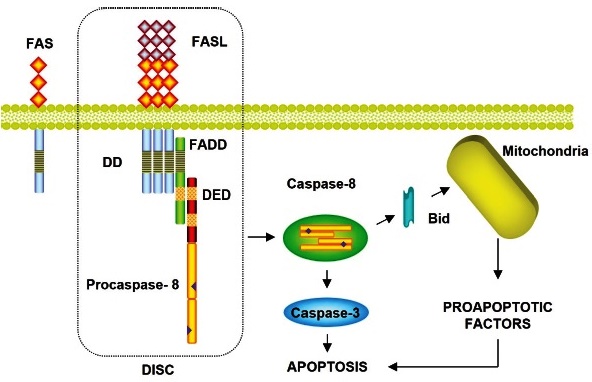
FAS is a cell surface receptor that, when binding to its ligand, causes apoptosis. (APO-1/CD95); FASL: Fas ligand; FADD: Fas Associated Death Domain; DD: Death Domain; DED: Death Effector Domain; DISC: Death-Inducing Signaling Complex; Bid: BH3 Interacting Domain Death Agonist Protein.
Diagram of apoptotic signals of the extrinsic pathway: mediated by FAS receptors of cell death. Caspase-8 can directly activate Caspase-3 or proteolyze the carboxy-terminal end of Bid, a proapoptotic protein of the Bcl-2 family. The translocation of the truncated form of Bid to the mitochondria will activate the mitochondrial pathway.
Fig. 2 Extrinsic apoptotic pathway by FAS cell death receptors.
The Fas / Fas ligand (FasL) system participates in the elimination of T and B lymphocytes, viruses-infected cells, and cancer cells. Doxorubicin and methotrexate (cytotoxic agents) or immunomodulatory drugs (IMiDs) activate this pathway to achieve cell death in malignant cells in cancer disease 37.
Furthermore, FasL can also interact with the DcR receptor, a soluble secreted receptor of the TNF superfamily. Some cells produce “decoy” receptors on the cell surface with a ligand-binding domain but no death domain; therefore, they can bind to a death ligand but do not trigger apoptosis. When FasL binds to DcR3, it inhibits FasL/ Fas apoptotic activity, thus acting as a “decoy.” Cells can also produce intracellular blocking proteins such as FADD-like IL -1β-converting enzyme (FLICE)-inhibitory protein (FLIP), which resembles procaspase but lacks a proteolytic domain; FLIP competes with procaspases-8 and 10 for binding sites on DISC and thus inhibits the activation of these initiating procaspases 38,39.
Tumor necrosis factor (TNF) receptor type 1
Tumor necrosis factor-α (TNF-α) is a type II transmembrane protein that mediates the inflammatory response, regulation of immune cells, and cytotoxicity. TNF-α binds to tumor necrosis factor receptor 1 (TNF-R1), also known as death receptor 1 (DR1), and tumor necrosis factor receptor 2 (TNF-R2). TNFR1 and TFNR2 are involved in pro-survival signaling and proliferation by activating the nuclear factor kappa B (NF-kB) pathway 31,40.
TNF-R1 can be found in all cell types, and TNF-R2 is mainly expressed in immune and endothelial cells 40. TNF-R1 is ubiquitously expressed, like Fas, whereas its ligand TNF-α is only expressed on activated macrophages and lymphocytes in response to infections 35. TNF-R1 has a death domain that can trigger apoptosis by activating the caspase cascade. Upon activation, TNF binds to its receptor via trimerization of TNFR1. Subsequently, a TNFR-associated death domain protein (TRADD) adapter molecule is added that induces association with FADD and activation of Caspase-8. In addition to the apoptotic pathway, TNF induces other signal transduction pathways from TRADD that trigger the activation of NF-κB and c-Jun Kinase (JNK)/Ap-1 41,42 (Fig. 3).
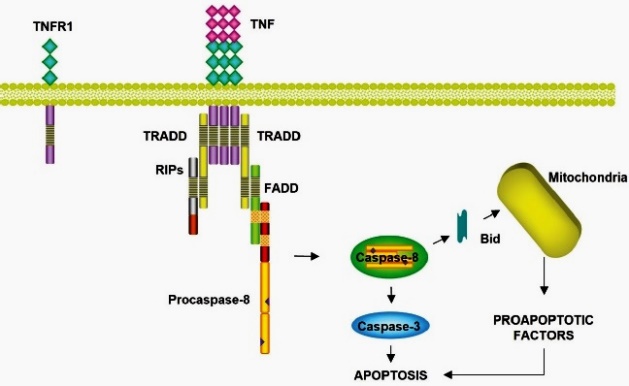
TNF: tumor necrosis factor; TNFR1: Tumor Necrosis Factor Receptor; TRADD: Tumor Necrosis Factor Receptor (TNFR)-Associated Death Domain protein; RIPs: Receptor Interacting Proteins; FADD: Fas Associated Death Domain; Bid: BH3 Interacting Domain Death Agonist Protein.
Signaling pathway of the TNF receptor 1. TRADD adapter molecule is added, which induces the association with FADD and the activation of Caspase-8.
Fig. 3 Extrinsic apoptotic pathway by Tumor Necrosis Factor Receptor type 1 cell death receptors.
TNF-Related Apoptosis-Inducing Ligand (TRAIL)
TRAIL is a type II homotrimeric transmembrane protein expressed on the surface of T cells, macrophages, and NK cells, modulating the immune response 31. Zinc binding is essential for recognizing the receptor and the subsequent induction of apoptosis by stabilizing the trimeric conformation in TRAIL residue Cys230, which is essential 43. When TRAIL binds to DRs, it induces receptor trimerization, which triggers the extrinsic apoptotic pathway in transformed cells without affecting non-transformed cells 44.
TRAIL binds to two death receptors (DR), DR 4 and DR5, and “three decoys receptors” (DcR) (DcR1, DcR2, and osteoprotegerin (OPG)). TRAIL -R1 or DR4 and TRAIL -R2 or DR5, with 60% homology, can trigger apoptosis and determine whether a cell is resistant or sensitive to TRAIL 31. DcR1, or TRAIL -R3, is a GPI-anchored protein lacking the intracellular and transmembrane domains, while DcR2 or TRAIL -R4 has an intracellular portion containing a truncated DD; both receptors are unable to induce apoptosis after binding of TRAIL 45,46. OPG is a soluble receptor that can be released from the cardiovascular system, gastrointestinal tract, lung, kidney, bone, and immune cells 47. OPG binds TRAIL and many ligands, including another member of the TNF family, the receptor activating nuclear factor kB ligand (RANKL) 48.
Intrinsic pathway: mitochondrial death pathway
Intrinsic apoptosis is activated from within the cell in response to injury or other forms of stress, such as DNA damage, lack of oxygen, UV radiation, nutrient or survival signals, oxidizing agents, drugs, and growth factors 49 (Fig. 4).
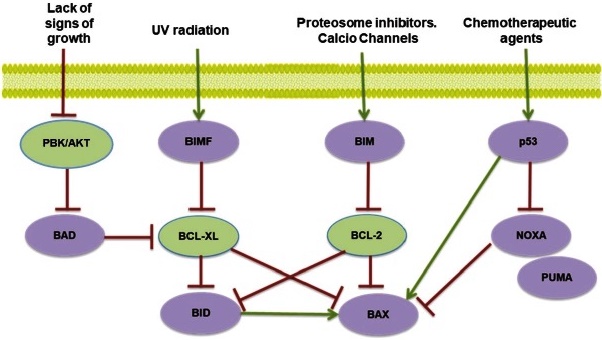
PBK: Protein kinase B; AKT: serine-threonine kinase (also known as protein kinase B (PKB) phosphorylated by PDK1 kinase.; BAD: Bcl-2 agonist of cell death; BIM: B-cell lymphoma 2 interacting mediator of cell death; BIMF: proapoptotic protein BIM family; BID: BH3-interacting domain death agonist; BCL -XL: B-cell lymphoma-extra-large; BCL -2: B-cell lymphoma 2; BAX: bcl-2-like protein 4; NOXA: (Latin for damage) is a proapoptotic member of the Bcl-2 protein family; p53: Tumor protein P53; PUMA: p53 upregulated modulator of apoptosis.
In signaling activation of the mitochondrial pathway of apoptosis, the BH3 domain is essential for apoptotic activity. Proteins that inhibit apoptosis and/or promote cell survival include Bcl-2, and Bcl-XL, located in the outer mitochondrial membrane, and the proteins Bim, Bad, Bid, Bimf, and Bax are found mainly in the cytosol and can be translocated to mitochondria in response to apoptotic stimuli.
Fig. 4 Potential signals of the activation of the intrinsic apoptotic pathway.
Although mitochondria were considered a passive element in apoptotic cell death, which only reflected damage to critical functions due to cell death 50, this apoptotic pathway depends on the release into the cytosol of mitochondrial proteins that normally reside in the intermembrane space 51.
Cytochrome-C
Cytochrome C is a protein that participates in the electron transport chain located in the intermembrane space of the mitochondria, and that can be used as a biomarker of the apoptosis process 1. Cytochrome C, performs an entirely different function; after being released into the cytosol, it binds to a procaspase-activating protein called apoptotic protease-activating factor-1 (Apaf1), causing the oligomerization of Apaf-1 into a heptameric wheel-like structure called apoptosome 52. In the apoptosome, Apaf1 proteins recruit initiator procaspase molecules (procaspase-9); these are activated into Caspase-9 and induce an apoptotic cascade activating one of the following chain executing procaspases-3, -6, and -7 inducing apoptosis53 (Fig. 5).
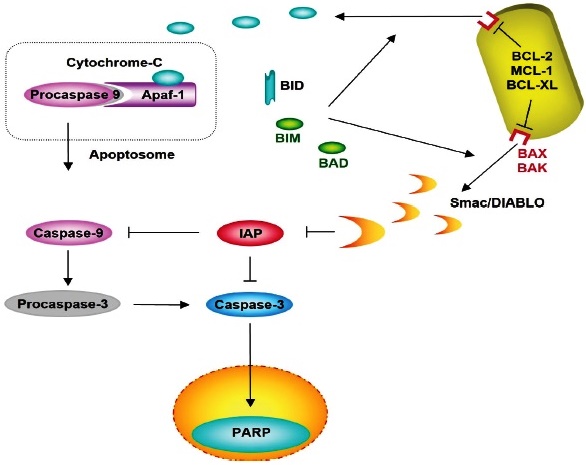
Apaf-1: Apoptotic protease-activating factor 1; IAP: Inhibitors of Apoptosis Protein; PARP: Poly (ADP-ribose) polymerase; BID: BH3 Interacting Domain Death Agonist; BIM: B-cell lymphoma 2 interacting mediator of cell death; BAD: Bcl-2 agonist of cell death; BCL -2: B-cell lymphoma 2; MCL -1: Induced myeloid leukemia cell differentiation protein; BCL -XL: B-cell lymphoma-extra-large; BAX: bcl-2-like protein 4; BAK: Bcl-2 homologous antagonist/killer; Smac / DIABLO: Second Mitochondria-derived Activator of Caspase.
Release to the cytosol binds to the Apaf-1 protein and procaspase-9, forming the apoptosome complex, inducing caspase-9 and the caspase activation cascade.
Fig. 5 Intrinsic pathway by Cytochrome C-mediated apoptosis.
Bcl-2 family proteins
Bcl-2 family of proteins controls and regulates the entire process of intrinsic apoptosis. Bcl-2 family proteins actively participate in this pathway, which, through interactions between them, regulates the permeabilization of mitochondria and the release of apoptogenic proteins into the cytosol 1. The Bcl-2 family of proteins contains at least one conserved domain, known as Bcl-2 homology domains (BH [BH1, BH2, BH3 and BH4]). The proteins of the Bcl-2 family are classified based on their function and structure: i) Antiapoptotic proteins, which contain the domains BH1 and BH2; ii) Proapoptotic proteins containing the BH1, BH2, and BH3 domains; iii) Proapoptotic proteins containing only the BH3 domain54. Most of the antiapoptotic members maintain sequence conservation in their four domains, while those with proapoptotic activity have less conservation of the first α-helix BH4 segment. Thus, the proteins of the Bcl-2 family are proapoptotic, and others are antiapoptotic. They can bind to each other in various combinations and form heterodimers in which the two proteins mutually inhibit each other. However, when a more significant proportion of these activities occur, the cell’s susceptibility to death or survival is determined 55,56.
Antiapoptotic proteins
Antiapoptotic Bcl-2 proteins, such as Bcl-2 itself, Mcl-1, Bcl-w, A1, and Bcl-XL, are found on the cytosolic surface of the outer mitochondrial membrane, endoplasmic reticulum, and nuclear envelope, where they help maintain membrane integrity. This group of proteins inhibits apoptosis and/or promotes cell survival 54. The three anti-apoptotic proteins of the Bcl-2 family, Bcl-2, Bcl-XL, and Mcl-1, prevent the activation of the mitochondrial apoptosis pathway, as demonstrated in multiple myeloma (MM) 37. In MM, the defect in the cell death pathways is frequently due to an imbalance in the expression of the Bcl-2 family proteins. The Bcl-2 gene has been implicated in resistance to apoptosis induced by dexamethasone but not melphalan in patients with MM. Bcl-XL is expressed in most MM cell lines and cells from patients; increased expression is frequently detected in the relapsed patient and correlates with resistance to chemotherapy. Mcl-1 is expressed in virtually all MM lines and fresh cells from patients. The induction of apoptosis in MM cells has been related to a decrease in the expression of Mcl-1 57. Also, for acute myeloid leukemia (AML), higher expression of Bcl-2, Bcl-XL, and Mcl-1 and lower expression of Bax increase resistance to apoptosis in CD34+ populations than in CD34-populations, mainly due to AML 58. Furthermore, increased expression of Bcl-2 and Bcl-XL blocks doxorubicin-induced apoptosis. Mcl-1 levels are increased in patients with recurrent AML 59.
Pro-apoptotic Bcl2 proteins
Proapoptotic Bcl-2 proteins comprise two subfamilies: the BH1-4 proteins that share four different homology domains (Bcl-2-associated X protein [Bax] and Bcl-2 homologous antagonist/killer [Bak] and the proteins with restricted homology to BH3. The BH3 domain is essential for apoptotic function 60. Among the members of the Bcl-2 family that induce apoptosis, with bounded homology to the BH3 domain, the following proteins are grouped: Bcl-2-interacting protein BIM (Bim), Bcl-2 agonist of cell death (Bad), Bid, Bcl-2 adenovirus E1B 19kDa- interacting protein 1 NIP3 (Bnip3), BMF, HRK, Noxa and p53 upregulated modulator of apoptosis (PUMA). These proteins are the largest subclass of the Bcl-2 protein family 61. Bak protein is tightly bound to the outer membrane of the mitochondria even in the absence of an apoptotic signal, whereas Bax is localized primarily in the cytosol and only translocates to the mitochondria if an apoptotic signal is activated. Bax and Bak activation depend on activated “BH3 one” pro-apoptotic proteins. Bax and Bak act on the endoplasmic reticulum’s and nuclear membranes’ surface; when activated in response to endoplasmic reticulum stress, they release Ca2+ from the endoplasmic reticulum into the cytosol. Bax and Bad are essential gateways for cell death through mitochondria 61. Restricted homology to BH3 proteins provides the crucial link between the apoptotic stimulus and the intrinsic apoptosis pathway; its BH3 domain binds to a long hydrophobic groove of the Bcl-2 antiapoptotic proteins and neutralizes or inhibits their activity. In some cells, the extrinsic apoptotic pathway recruits the intrinsic pathway by amplifying the caspase cascade that kills the cell. In this way, Bid is the link between the two pathways. Also, Bid, Bim, and PUMA can inhibit all Bcl-2 antiapoptotic proteins 62,63. Klee et al.64 have investigated mitochondrial membrane permeabilization, which depends not only on the canonical mitochondrial Bak and Bax pathway to activate the death program. Therefore, these investigators have also found that the “only-BH3” molecules, Bim and PUMA, can induce the release of cytochrome c and apoptosis with the mere presence of Bak in the endoplasmic reticulum 64. This pathway for transmitting apoptotic signals from the endoplasmic reticulum to mitochondria involves coordinated communication mediated by the calcium and ER1α/TRAF2 ER-stress surveillance machinery 61,63,65.
Second Mitochondria-derived Activator of Caspase (Smac / Diablo)
Second mitochondria-derived activator of caspase (Smac / Diablo) binds by its N-terminal end to the mitochondria and, in the intermembrane space, proteolyzes, leaving free the domain that allows its union with the IAPs 66. The loss of mitochondrial potential simultaneously triggers the release of cytochrome-C and Smac/Diablo. Smac/Diablo, in the cytoplasm, is capable of binding to IAPs (XIAP, c-IAP1, c-IAP2, and survivin), inhibiting their function and enhancing caspase activation and triggering the mitochondrial apoptosis pathway 67. The release of Smac / Diablo is inhibited by Bcl-2 and Bcl-xL 54. In MM cells, Smac plays a functional role in mediating the activation and apoptosis of Caspase-9 induced by dexamethasone treatment 37.
Caspase-independent pathways of apoptosis
Mitochondrial caspase-independent pathway
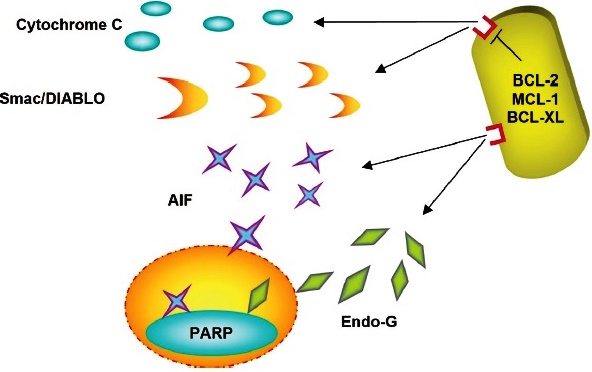
The loss of mitochondrial potential increases the permeability of the mitochondrial membrane, and the result is the release of these proteins, such as AIF (apoptosis-inducing factor) and Endonuclease G (Endo-G) that activate a caspase-independent apoptosis pathway 68.
AIF (Apoptosis Inducing Factor)
Apoptosis-inducing factor (AIF) is a highly phylogenetically conserved protein, essential for embryonic development, which is synthesized in the form of an immature precursor; it is translocated to the mitochondria, and in the intermembrane space it is proteolyzed, the mature form has oxidase activity 69. In response to death signals, AIF leaves the mitochondria and travels through the cytosol to the nucleus, where it binds to DNA, causing chromatin condensation and DNA fragmentation into fragments of approximately 50 kb 16 (Fig. 6). AIF is necessary to induce PARP-dependent death. The processing and activation of PARP occur in response to DNA damage. PARP initiates a signal in the nucleus that induces the release of AIF from the mitochondria. AIF then moves from the mitochondria to the nucleus and induces chromatin condensation and DNA fragmentation 69,70.
RIP3: Receptor-interacting serine/threonine-protein kinase 3; RIP1 Receptor-interacting serine/threonine-protein kinase 1; FLIPs: FLICE-inhibitory proteins; FAAD: Fas-associated death domain protein.
In response to death signals, AIF leaves the mitochondria and moves through the cytosol to the nucleus, where it binds to the DNA, causing chromatin condensation and DNA fragmentation.
Fig. 6 Proapoptotic factors as Cytochrome-C, Smac/Diablo, Endonuclease, and Apoptosis Inducing Factor are released from the mitochondria.
Endonuclease G (Endo-G)
Endonuclease G (Endo-G) is an essential protein for mitochondrial DNA replication. It was isolated from the mitochondrial fraction treated with the proapoptotic active form of Bid: tBid. Once released to the cytosol, it is transferred to the nucleus, where it fragments DNA, even in the presence of caspase inhibitors (Fig, 6). Endo-G cooperates with exonuclease and DNase, facilitating DNA processing 71,72. Apoptotic endonuclease acts cooperatively to fragment DNA and ensure the irreversibility of apoptosis. However, very little is known about the potential regulatory linkages between endonucleases. Therefore, deoxyribonuclease deactivation is caused by cutting. Also, alternative splicing of DNase I pre-mRNA skipping exon 4 occurs in response to overexpression of Endo-G in cells 16,72. Likewise, a strong correlation was identified between the expression levels of Endo-G and DNase I splice variants in human lymphocytes. In fact, T cells downregulate the mRNA levels of the active full-length DNase I variant. They also upregulate the levels of the inactive spliced variant, which acts in a dominant-negative fashion 70,71.
Necroptosis
Necroptosis is a form of programmed cell death since it is genetically regulated; it is characterized by cell inflammation, mitochondria dysfunction, plasma membrane permeabilization, and the release of cytoplasmic content into the extracellular space, causing inflammatory reactions in the cells of the surrounding tissue. This form of cell death is also associated with mitochondrial reactive oxygen species (ROS) and, unlike apoptosis, does not involve DNA fragmentation 73. Necroptosis has been reported to occur in a wide range of human diseases, including retinal ischemia-reperfusion injury, acute pancreatitis, brain trauma, retinal detachment, and Huntington’s disease 73,74.
In addition, necroptosis has been linked to models of inflammation, including intestinal inflammation and systemic inflammatory response syndrome (SIRS) 75,76. Perhaps the detailed knowledge of this cell death pathway can be used to develop drugs that temporarily prevent or block this process to delay the death of some types of cells or, conversely, it could also serve to eliminate selectively, for example, tumor cells 77.
Activation Pathways of Necroptosis
Necroptosis is triggered as a form of immunity against pathogens, under poor conditions to trigger apoptosis. In necroptosis, as in apoptosis, TNF activates TNFR1, which induces the activation of a serine/ threonine kinase interaction protein (RIP1), making integrating a joint inflammatory and necroptotic response possible 78. RIP3 is activated upon phosphorylation by the serine/threonine kinase RIP1 79. Necroptosis is RIP3-dependent as RIPK3 protein kinase activity determines whether cells die by apoptosis or necroptosis. Perhaps necroptosis is fundamentally characterized by the activation of RIP1 or RIPK3, while the caspase cascade is inhibited 79. Giampietri et al.80 have proposed a model that differentiates the production of cell death by apoptosis and by necroptosis; the dimerization of Caspase-8 produces apoptosis while in necroptosis it does not occur. The dimerization of C-Flip S and caspase-8 produces a reduction in caspase-8 activity, and it may not produce either apoptosis or necroptosis, finally, the heterodimerization of C-FLIPs and Caspase-8 produces inhibition of Caspase-8 and leads to the production of necroptosis (Fig. 7).
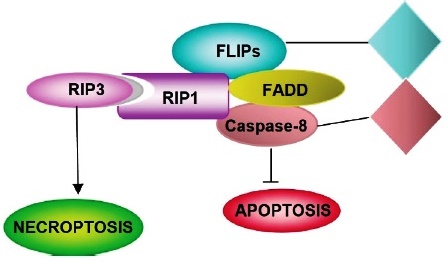
RIP3: Receptor-interacting serine/threonine-protein kinase 3; RIP1 Receptor-interacting serine/threonine-protein kinase 1; FLIPs: FLICE-inhibitory proteins; FAAD: Fas-associated death domain protein.
Necroptosis signaling pathways mainly comprise heterodimerization to c-FLIPs that reorganize the catalytic site procaspase-8, producing caspase-8 inhibition, which induces necroptosis.
Fig. 7 Pathway of specific activation of necroptosis.
Regulation Pathways of Necroptosis
Liu et al. 81 have shown that the Akt and mTOR pathways regulate necroptosis by inducing RIPK1 activation in neuronal cell death. Just as it has also been verified that necrostatin-1 is an inhibitor of all the biochemical events carried out in this type of cell death. In addition, other investigators 82 have reported that necroptosis is paired with a mixed lineage kinase domain-like protein (MLKL) gene, an important substrate of RIP3, and the plasma surface pores are constituted by said protein (Fig. 8). These pores cause the absorption of too much water, so the cells ultimately burst. The blockade of MLKL activity leads to the inhibition of necroptosis 82. In this sense, Dondelinger et al.82 have proposed that a domain of four activated MLKL molecules is required to induce its oligomerization and trigger cell death.
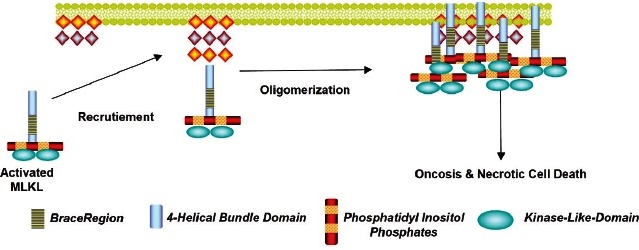
MLKL: Mixed Lineage Domain-Like Protein Kinase.
The MLKL protein has become a specific and crucial protein. The 4-Helical Bundle Domain (4HBD) in the N-terminal region of MLKL is required to induce its oligomerization. and trigger cell death.
Fig. 8 Recruitment of Mixed Lineage Domain-Like Protein Kinase by Phosphatidylinositol. Pathway to the plasma membrane.
On the other hand, it has been found that phosphatidylinositol (PIP) recruits the MLKL protein to the plasma membrane. Of note, recombinant MLKL lacks positive charges and induces leakage of liposomes containing both PIP and BAX, supporting a model in which MLKL induces necroptosis by directly permeabilizing the plasma membrane. Consequently, inhibition of PIP formation specifically inhibits TNF mediated by necroptosis but not apoptosis 82.
Autophagy
The term autophagy was introduced in 1996 by De Duve and Wattiaux, who defined the vacuolization process for transporting intracellular material to lysosomes for degradation 83. Autophagy is derived from the Greek auto and phagos; it literally means “self-feeding.” Its function mainly regulates intracellular homeostasis since cytoplasmic materials (long-lived proteins and damaged organelles) are degraded in lysosomes and recycled to produce new building blocks and maintain energy metabolism 84. From a morphological point of view, autophagy has been classified as a form of programmed cell death associated with the massive accumulation of autophagosomes in the cytoplasm, which frequently, but not always, seems to be accompanied by increased blood flow; massive autophagy triggers caspase-independent, necrosis-like death 85. It has been shown that autophagy participates in natural processes such as growth, embryonic development, or aging. Also, it participates in the death that occurs in mammary cells after lactation and the death of some cancer cells that lack apoptotic modulators, such as Bax and Bak or caspases 85,86. Dysfunction of this process has been linked to cardiovascular and respiratory diseases, neurodegenerative diseases, metabolic diseases, and cancer 86,87.
Autophagy is a type of programmed cell death since more than 30 genes have been identified in yeast that regulate autophagy, and it is seen as a survival mechanism to combat environmental stress factors 88. Autophagy could be induced in response to oxidative or metabolic stress and can also be induced through starvation, which is very commonly used in research 86. On the other hand, to demonstrate that cell death in an in vivo or in vitro model is caused by autophagy, it is necessary to demonstrate that said death is inhibited by agents interrupting the autophagic pathway 88. These agents can be chemical (agents directed against VPS34), genetic (siRNA 3-methyl-adenine), or modulators of autophagy (AMBRA 1, ATG 5, beclin) 89.
Types of autophagy
Three types of autophagy are identified: i) macroautophagy; ii) microautophagy; iii) chaperone-mediated autophagy 84. The macroautophagy process, as described by Kwanten et al. 88 begins with the formation of a phagophore, a double membrane structure (also known as an isolation membrane) that sequesters cytoplasmic material (long-lived proteins and organelles), which subsequently elongates to create an autophagosome. The autophagosome fuses with a lysosome to form an autolysosome, where its contents will be degraded by lysosomal proteases (e.g., cathepsins) and other hydrolytic enzymes 90,91. According to Kwanten et al. 88, phagophore formation is regulated by the ULK1 complex (initiation), which is under the control of the mammalian target of rampamycin (mTOR) complex and the beclin-1/ VSP34 interaction complex (nucleation). Two large, conjugated ubiquitin-like complexes are responsible for double membrane elongation: light chain 3 (LC3)-II and ATG5-Atg12- ATG16L1. ATG7 is one of the proteins required to form both elongation complexes. Autophagosomes are generated on or in the vicinity of the endoplasmic reticulum. However, it is not clear whether the ER membrane is used directly for autophagosome formation. Recent studies suggest that additional membranes derived from the Golgi complex, mitochondria, and the plasma membrane also contribute to autophagosome formation 90,91. Therefore, autophagosome formation involves multiple and complex processes 84. Macroautophagy is considered to play the most critical role in autophagy 92 (Fig. 9).
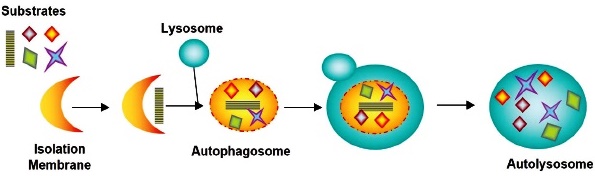
In the autolysosome, the inner membrane and luminal content of the autophagic vesicle are degraded by lysosomal enzymes that act optimally within this acidic compartment.
Fig. 9 Macroautophagy process from isolation membrane to autolysosome.
Microautophagy process is considered when a small portion of the cytoplasm is directly involved by the lysosome / late endosome (Fig. 10). Autolysosome formation is mediated by the accumulation of small membrane structures that envelop portions of the cytoplasm. Phagophores are not formed, and membrane structures invaginate directly into lysosomes, where degradation occurs by direct absorption of cytoplasmic cargo.
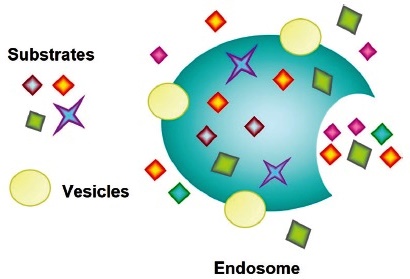
Microautophagy is conserved from yeast to mammals and contributes to the degradation of organelles (e.g., peroxisomes, ER, nucleus), protein complexes such as the proteasome, and single proteins.
Fig. 10 Endosomes result from the microautopha- gy process.
In Chaperone-Mediated Autophagy (CMA), the proteins to be degraded are delivered selectively to lysosomes; they are recognized by heat shock-like proteins 70 (HSC70) and by co-chaperones; proteins in the degradation phase are internalized through lysosomal membrane-associated protein 2A (LAMP2A) 91,92 (Fig. 11).
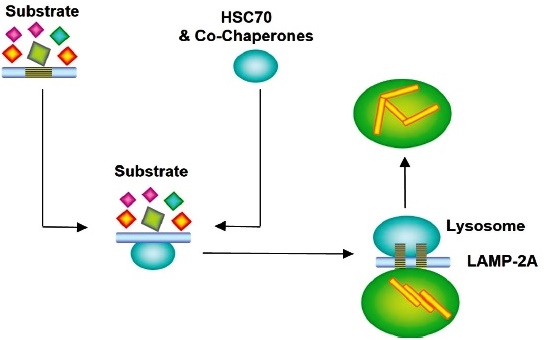
HSC70: Heat shock 70 kDa protein cognate; LAMP-2A: Lysosome-associated membrane protein 2A.
The proteins to be degraded are delivered selectively to lysosomes, which HSC70 and co-chaperones recognize. Proteins in the degradation phase are internalized through LAMP2A.
Fig. 11 Lysosome obtained from Chaperone-Mediated Autophagy pathway.
Autophagy: a selective process
Degradation in the autophagy process was believed to be non-selective; however, it has been determined that there are selective pathways to digest specific components, such as “mitophagy” or selective autophagy of mitochondria 93, “peroxyphagy” (peroxisomes), “ribophagy” (ribosomes) or “xenophagy” (invading microbes), this phenomenon is called selective autophagy, and in the case of mitochondria, it serves to maintain their homeostasis 70,84. Thus, macroautophagy can be non-selective (random uptake of intracellular material) and selective (specific load capture). The morphological and biochemical characteristics of autophagy and apoptosis are different. In this regard, cells undergoing autophagy show an increase in autophagic vesicles (autophagosomes and autophagolysosomes). While chromatin condensation is partial in autophagic cells, DNA fragmentation does not occur. The two processes, autophagy and apoptosis, are not always mutually exclusive and can occur simultaneously in the same type of cells 91.
In summary, homeostasis is maintained between cells produced by mitosis and cell death in the human body. In this sense, this study is a narrative review that reports scientific research in which an attempt has been made to group programmed cell deaths, explaining the molecular mechanisms that involve structural and functional proteomic pathways that intervene by inducing and inhibiting each one of the proteomic pathways. In our study, caspase-dependent programmed cell deaths and caspase-independent programmed cell deaths are described. Although classifying and describing programmed cell death processes is somewhat complex, depending on which aspects are analyzed, their grouping and knowledge of the factors that trigger cell death vary greatly. This study could offer the basis for the design of new pharmacological treatments and discover new potential molecular biomarkers for early diagnosis that serve to cure or modulate the course of some diseases. For this, it is necessary to understand the proteomic signaling mechanisms of programmed cell death since their alteration contributes to a wide variety of diseases, one of which is cancer, which constitutes a global public health problem due to its high mortality.